
Ansgar Heck.
Av Ansgar Heck, stipendiat, Seksjon for spesialisert endokrinologi, OUS, Rikshospitalet
Akromegali er en sjelden sykdom som i tidlig sykdomsfase har uspesifikke symptomer. Derfor stilles diagnosen gjerne med mange års forsinkelse og etter at irreversible skjelettforandringer og organkomplikasjoner er etablert. Stilles diagnosen tidlig øker sjansen for helbredelse, og de irreversible forandringene er mindre uttalt. Dette forutsetter at man kjenner til de uspesifikke symptomene, har sykdommen i mente og foretar målrettet prøvetaking hvis man møter en pasient med kombinasjon av flere uspesifikke symptomer.
Akromegali er en sjelden sykdom som nesten utlukkende er forårsaket av et veksthormonproduserende hypofyseadenom. Insidensen ligger på rundt 0,4/100 000/år [1]. I Norge kan man altså forvente ca. 20 nye tilfeller per år. Antakeligvis finnes det mellom 500-800 pasienter med akromegali her i landet.
Hos friske er veksthormonverdier lave gjennom store deler av døgnet, med kortvarige topper først og fremst om natten og i forbindelse med faste og stress. Veksthormon stimulerer hepatisk produksjon av IGF-1 (Insulin like growth factor 1) som kan måles i serum. Mange veksthormoneffekter medieres via systemisk IGF-1, men delvis også gjennom lokal IGF-1 produksjon (parakrin effekt). Direkte veksthormoneffekter er lipolyse i fettvev på den ene siden og økt triglyseridopptak i lever og muskel på den andre siden [2].
I motsetning til hos friske ser man ved akromegali vanligvis et kontinuerlig forhøyet veksthormonnivå (se figur 1) og gjennom dette økt IGF-1 nivåer.
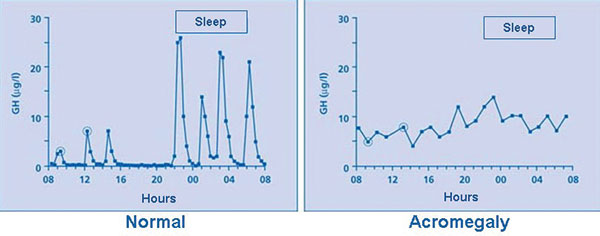
Figur 1: Typisk veksthormonprofil hos friske med kortvarige sekresjonstopper om natten og ved faste (venstre side), og kontinuerlig forhøyet veksthormonsekresjon hos en pasient med akromegali (høyre side) [20]; Gjengitt i henhold til BioMed Central Open Access license agreement.

Tabell 1.
Symptomene kan inndeles i klassiske, ofte irreversible forandringer på den ene siden, som gjerne består av skjelettforandringer og leddsmerter, og på den andre siden uspesifikke, reversible symptomer, som f. eks. svetting, hevelse i fingre og tær, hodepine og hypertensjon (tabell 1). Forandringene som har gitt sykdommen navnet, utvikler seg over lang tid og består av karakteristiske skjelettforandringer som breddeøkning av hender og føtter, forandringer i ansiktsskjelettet med vekst av kjeve, kinnbein og pannebein.
Leddsmerter forårsaket av akromegal arthropati påvirker livskvaliteten hos mange pasienter med akromegali, likner klinisk på arthrosesmerter, men mangler ofte røntgenologiske slitasjetegn som avsmalnet leddspalte [3].
Den direkte lipolytiske virkningen av veksthormon fører til at pasienter med akromegali ofte er slanke og vanligvis ikke har abdominal fedme. Direkte veksthormoneffekter på lever og skjelettmuskel fører til at pasienter med akromegali ofte er insulinresistente og kan ha nedsatt glukosetoleranse eller manifest diabetes selv om de er slanke.
Bløtdelshevelse kan forsterke inntrykket av store hender og føtter og kan føre til typiske komplikasjoner som karpaltunnelsyndrom og obstruktivt søvnapnesyndrom.
Utfordringen er å stille diagnosen akromegali tidlig i den store gruppen av personer med hyppig forekommende, uspesifikke symptomer (tabell 1).
Nøkkelen til en vellykket behandling ligger i å stille diagnosen før pasienten får irreversible symptomer. Dette øker også sjansen for definitiv biokjemisk helbredelse etter transsfenoidal tumorreseksjon. Pasienter som ikke kan helbredes ved kirurgisk behandling, kan trenge langvarig medikamentell behandling eller eventuell strålebehandling. Problemet med å stille diagnosen tidlig er at symptomene i denne sykdomsfasen er uspesifikke, krever årvåkenhet fra legenes side og målrettet prøvetaking. Mangel på eller lite uttalte akromegale trekk utelukker ikke diagnosen (figur 2). Tvert imot bør man være liberal med å tenke på diagnosen hvis en pasient har to eller flere uspesifikke symptomer (se tabell), også hvis typiske stigmata ikke fremtrer tydelig. Spørsmål om økt ringstørrelse og skostørrelse (bredde) kan bidra til å identifisere pasienter som bør vurderes for prøvetaking (IGF-1).
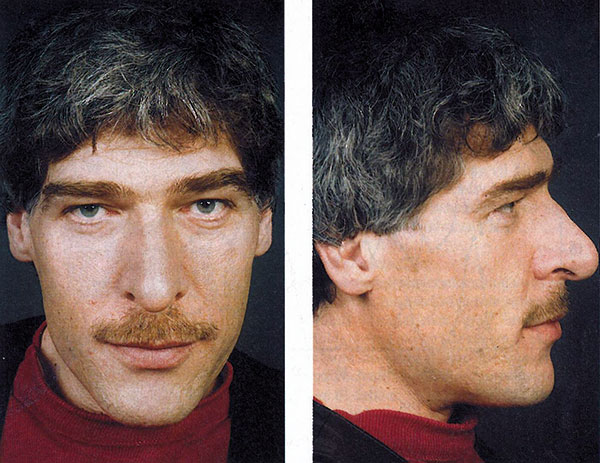
Figur 2: Diskré, men typiske trekk hos nydiagnostisert 42-årig mann med akromegali. Bildene er gjengitt med pasientens og øvrige tillatelser [13].
Bestemmelse av IGF-1 er det første utredningstrinnet om det foreligger mistanke om akromegali. De største sykehuslaboratoratorier i Norge har analysen i sitt repertoar (Hormonlaboratoriet Aker, Rikshospitalet, Haukeland sykehus, St. Olavs Hospital, Universitetssykehuset Nord-Norge (UNN)). Referanseområdet er aldersavhengig. Prøven må sentrifugeres innen 30 min etter prøvetaking og sendes på is. Dette kan by på utfordringer i primærhelsetjenesten, men er vanligvis uproblematisk hvis prøve tas på et sykehuslaboratorium.
Siden veksthormonsekresjonen skjer pulsatilt hos friske opererer man ikke med et avgrenset normalområde for veksthormon. Dermed har enkeltbestemmelse av veksthormon ingen diagnostisk verdi ved mistanke om akromegali [4].
Diagnosen sikres ved standard oral glukosebelastning, og denne testen benyttes også for å avgjøre om pasienten har aktivitet i sykdommen etter kirurgisk behandling [5].
Hos friske vil en standardbelastning med 75 g glukose supprimere veksthormonkonsentrasjonen til mindre enn 2,5 mU/l, mens dette ikke er tilfellet ved akromegali. Det diskuteres om denne grensen bør senkes til 1 mU/l [4].
Som ledd i basisutredning bør det tas hypofyseprøver (tabell 1) for å avdekke eventuell hypofysesvikt eller koproduksjon av andre hypofyseforlappshormoner, som f. eks. prolaktin (hos ca. 25 %) eller TSH (meget sjeldent).
Radiologisk verifisering og kartlegging av tumor skjer fortrinnsvis ved MR-skanning (figur 3). Veksthormonproduserende hypofyseadenomer går vanligvis ut fra den laterale delen av hypofyseforlappen. Mindre adenomer ligger vanligvis intrasellært, mens makroadenomer (størrelse over 10 mm) strekker seg ofte lateralt ut mot sinus cavernosus. De preoperative bildene vil ofte indikere om tumor lar seg radikaloperere, og har derfor stor prognostisk verdi [6]. Hvis adenomet vokser kranialt, kan dette føre til affeksjon av chiasma opticum og gi synsforstyrrelse. Om det foreligger klinisk synsaffeksjon og radiologisk chiasmaaffeksjon, bør utredning og behandling skje rask.
T2 vektede MR bilder kan bidra til å forutsi medikamentell behandlingseffekt [7, 8].
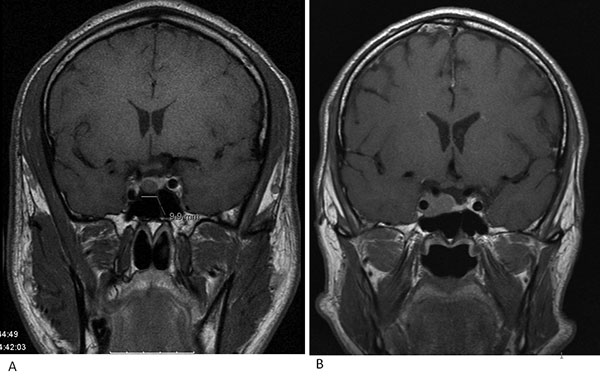
Figur 3: MR av hypofysen av to pasienter med veksthormonproduserende hypofyseadenomer. Til venstre (A) foreligger det et mikroadenom (første diameter under 1 cm). Det foreligger en god sjanse for biokjemisk remisjon etter transsfenoidal hypofyseoperasjon. Til høyre (B) et makroadenom med invasjon i høyre sinuscavernosus. Biokjemisk remisjon kan ikke forventes etter transsfenoidal hypofyseoperasjon.
Vanlige eller typiske manifestasjoner og komplikasjoner er hypertensjon, obstruktivt søvnapnesyndrom, nedsatt glukosetoleranse eller manifest diabetes mellitus. Disse er helt eller delvis reversible hvis man oppnår biokjemisk kontroll på akromegalien. Behandlingen av komplikasjonene skjer etter vanlige kliniske retningslinjer. Akromegali er forbundet med en lett økt relativ risiko for forekomst av kolonpolypper og koloncancer [9, 10]. Det anbefales derfor koloskopi rundt diagnosetidspunktet [5]. Det bør undersøkes om det foreligger kardiomegali ved ekkokardiografi. Om det foreligger bittforandringer bør dette kartlegges og evt. behandles av tannlege eller kjeveortoped (f. eks tannhelsekompetansesenter, www.tako.no).
Pasienter med ubehandlet akromegali har økt mortalitet og morbiditet. Ved å oppnå biokjemisk kontroll på sykdommen kan man redusere mortaliteten til nivået i bakgrunnsbefolkningen [11]. Det er mulig å bedre morbiditeten, de metabolske forandringene og de reversible symptomene. Skjelettforandringene er irreversible og leddsmertene har tendens til å persistere.

Standardbehandlingen ved akromegali er transsfenoidal operasjon med reseksjon av den veksthormonproduserende tumor. Inngrepet kan utføres med begrenset risiko, og alvorlige komplikasjoner er sjeldne [12]. Ved operasjon av hormonproduserende hypofyseadenomer må muligheten til å fjerne mest mulig av adenomet avveies mot økt risiko for komplikasjoner, blant annet hypofysesvikt.
På diagnosetidspunktet er kun 7 – 20 % av hypofyseadenomene mikroadenomer (< 10 mm) [6, 13]. Remisjonsraten for ikke-invasive makroadenomer ligger på 40-68 % [5], mens invasive makroadenomer har en betydelig lavere postoperativ remisjonsrate, sannsynligvis på < 10 % [6, 14].
Primær behandling med somatostatinanaloger kan vurderes i de tilfellene hvor man ikke kan forvente kirurgisk helbredelse [5]. Etter ½ – 1 år forbehandling tar man så på nytt stilling til om det skal utføres hypofysekirurgi. Pasienter som ikke helbredes ved kirurgi må vanligvis regne med å stå på livslang medikasjon.

Tabell 2.
Somatostatinanaloger
Hovedpilaren i den medikamentelle behandlingen er somatostatinanaloger. Octreotide og lanreotide er de i øyeblikket kommersielt tilgjengelige medikamenter. Medikamentene foreligger i formuleringer som gir forsinket frigjøring og injiseres vanligvis hver fjerde uke, lanreotide kan også gis med lengre intervaller (opptil syv uker).
Medikamentell behandling med somatostatinanaloger kan gis som primær behandling eller som adjuvant behandling av pasienter som ikke ble helbredet ved kirurgi.
Hos omtrent ¼ av pasientene får man ikke kontroll på veksthormonnivåene med til tross for maksimal dosering med somatostatinanaloger. Octreotidetest og T2 vektet MR kan bidra til å predikere effekten av somatostatinagonistene [8]. Hos pasienter hvor man ikke oppnår kontroll med veksthormonsekresjonen, må det vurderes ny operasjon, strålebehandling eller kombinasjonsbehandling med dopaminagoister eller veksthormonantagonister.
Om kort tid er det forventet at en ny somatostatinanalog (Pasireotide) markedsføres i Norge, men det må avventes resultatene fra pågående studier før dens plass i den kliniske behandlingsalgoritmen kan avklares.
De fleste tåler somatostatinanaloger bra. De vanligste bivirkningene er gastrointestinalt besvær, med oppblåsthet, diaré og abdominalsmerter [15].
Dopaminagonister
Dopaminagonister (f. eks. cabergoline eller bromocriptin) har effekt hos cirka 1/3 av pasientene, noe mer ved svulster som også produserer prolaktin (co-produksjon). Behandlingen kan gis som monoterapi, eller i kombinasjon med annen medikamentell behandling [15]. Gastrointestinale bivirkninger, hodepine, trøtthet og hypotensjon er vanlige, men ofte forbigående. Psykiske bivirkninger forekommer. Ved bruk av cabergoline i høye doser over lang tid kan det sees fibrotiske forandringer på hjerteklaffene. I doser som er vanlig ved endokrinologiske indikasjoner ser man ingen vesentlig økt forekomst av denne komplikasjonen, men det anbefales likevel fortsatt kontroll med ekkokardiografi [16, 17].
Veksthormonantagonister
Det nyeste medikamentelle behandlingsalternativet er veksthormonantagonister (pegvisomant). I motsetning til somatostatinanaloger og dopaminagonister nedsetter veksthormonantagonister ikke veksthormonproduksjonen på hypofysenivå, men motvirker veksthormoneffekten i perifere vev. Veksthormonantagonismen i leveren fører til lavere systemisk IGF-1. Behandlingen med pegvisomant må settes som daglig selvinjeksjon, men mange pasienter klarer seg med sjeldnere injeksjon, f. eks. 2 ganger per uke). Behandlingen er vanligvis reservert til pasienter hvor man ikke kommer i mål med annen medikamentell eller kirurgisk terapi, siden medikamentet er dyrt, og det kun foreligger begrenset langtidserfaring. Pegvisomant gis ofte som kombinasjonsbehandling med somatostatinanaloger.
Strålebehandling:
Stereotaktisk strålebehandling eller gammakniv, er i dag kun unntaksvis aktuelt og må avveies mot øvrige annenlinjes behandlingsmodaliteter. Med flere og bedre tilgjengelige medikamentelle behandlingsalternativer har andelen pasienter hvor strålebehandling er aktuelt sunket betydelig. Ulempen med strålebehandlingen er latenstiden før virkningen inntrer, og stor risiko for gradvis utvikling av hypofysesvikt [18].
Klarer man å få kontroll på veksthormonsekresjonen synes leveutsiktene å være på linje med normalbefolkningen [11]. Livskvaliteten er i stor grad avhengig av tilstedeværelsen av irreversible symptomer som f. eks. skjelett- og leddforandringer [19]. Sjanse for permanent biokjemisk remisjon er avhengig av adenomstørrelse og invasivitet.
Tidlig diagnose er viktig fordi den øker sjansen for vedvarende remisjon. Utfordringen ligger i at symptomene i tidlig sykdomsfase er uspesifikke. Foreligger det flere uspesifikke symptomer bør diagnosen akromegali vurderes selv om det ikke foreligger typiske skjelettforandringer. Årvåkenhet hos leger i primærhelsetjenesten, spesialisthelsetjenesten og spesielt indremedisinere, er avgjørende for pasientens livskvalitet og et godt behandlingsresultat. IGF-1 er best egnet som initial utredningsprøve.



