
Astrid Olsnes Kittang.
Av Astrid Olsnes Kittang, konst. overlege PhD, Hematologisk seksjon, Medisinsk avdeling, Haukeland Universitetssykehus
Myelodysplastiske syndromer består av en rekke sykdommer med patologi i benmarg, som alle kjennetegnes med en eller flere cytopenier i blodet. Artikkelen gir en kort beskrivelse av sykdommen og redegjør for moderne diagnostikk og behandling.
Myelodysplastiske syndromer (MDS) er en gruppe klonale stamcellesykdommer med vesentlig morbiditet og mortalitet. Det er store variasjoner i alvorlighetsgrad av sykdommen mellom pasientene, og pasientene kan ha svært ulik prognose. I dag benyttes et internasjonalt scoring-system for MDS som deler pasientene inn i fire risikogrupper på bakgrunn av antall cytopenier, prosent blaster i benmargen og cytogenetikk. Hvilken risikogruppe pasienten befinner seg i er av avgjørende betydning for legens videre behandling og oppfølging av pasienten.
Myelodysplastiske syndromer (MDS) ble tidligere kalt preleukemi og er en gruppe benmargsneoplasier med øket risiko for å gå over i akutt myelogen leukemi (AML). Sykdommen er klonal og dens utgangspunkt er postulert å være på nivået av hematopoietiske stamceller. Karakteristisk for MDS er cytopeni samt morfologiske forandringer i cellene som defineres som dysplasi i minst 10 % av cellene i en eller flere av de hematopoietiske cellerekkene. Forandringene kan være ledsaget av en økning av umodne myeloide celler i benmargen til 5 % blaster eller mer, men må ikke overskride 20 %.For en detaljert presentasjon av de morfologiske forandringer i blod og benmarg viser man til nettadressen http://imagebank.hematology.org/ som er linken til ASH (American Society of Hematology) Image Bank.
Det er en viss overhyppighet av sykdommen blant menn, og med en median insidensalder på 70 år, må man si at det først og fremst er en sykdom som rammer de eldre. Det er likevel viktig å huske på at MDS kan oppstå i alle aldersgrupper. Den årlige insidensen ligger på omkring 4 per 100 000. Sykdommen oppstår vanligvis uten noen kjent årsak, men den forekommer også i kjølvannet av annen hematologisk sykdom eller som langtidskomplikasjon av standard kjemo- eller radioterapi. Sannsynligvis er MDS i dag en underdiagnostisert lidelse og insidensen er også forventet økende med stadig bedre langtidsoverlevelse etter kreftbehandling.
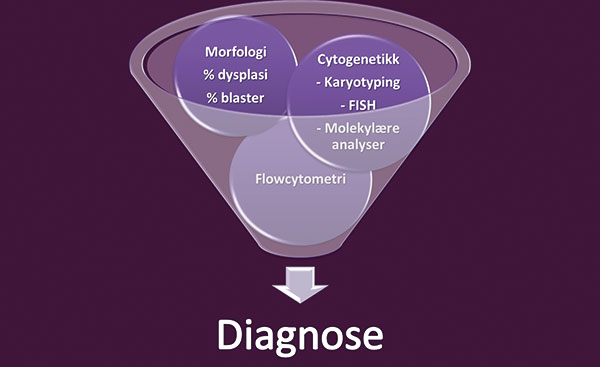
Figur 1. Diagnostiske hjelpemidler. I tillegg til hematologiske blodprøvesvar, hviler MDS-diagnostikk i dag på morfologi og cytogenetikk. I fremtiden vil trolig flowcytometriske parametre også inngå i diagnostikken.
MDS kan være en vanskelig diagnose å forholde seg til for pasientene. Som malign benmargsneoplasi må tilstanden meldes til Kreftregisteret. Hos yngre pasienter er allogen benmargstransplantasjon et kurativt behandlingstilbud, men de fleste pasientene er på grunn av alder og/eller komorbiditet ikke kandidater for dette og vil bare tilbys palliativ behandling. Hos mange av lavrisikopasientene med lav hemoglobin vil det være indisert å starte med vekstfaktorbehandling, noe som kan virke ulogisk i en neoplastisk sammenheng. En god del av pasientene vil kun tilbys støttende behandling eller sågar observeres uten noen som helst behandling og kan leve med en lavrisikosykdom i flere tiår. Hyppige blodtransfusjoner er vist å kunne forkorte overlevelsen [1], og hos enkelte vil det være behov for jernchelerende behandling for å redusere ferritinnnivået. Pasientene kan også ha autoimmune manifestasjoner, som vil kunne kreve behandling for eksempel med lokale eller systemiske sterioder. Det er sett økt binding av metylgrupper på arvestoffet hos pasienter med MDS, noe som kan føre til at viktige tumorsuppressorgener ikke blir avlest. Hos høyrisikopasienter vil hypometylerende behandling kunne normalisere perifere blodverdier og forlenge overlevelsen [2]. Alvorlige infeksjoner og blødninger er vanlige komplikasjoner og dødsårsaker for MDS-pasientene, i tillegg til utvikling av akutt leukemi. Således kan myleodysplastisk sydrom være utfordrende også for indremedisineren, men det finnes en del hjelpemidler i litteraturen, i tillegg til nasjonalt handlingsprogram for maligne blodsykdommer, som er på trappene, og retningslinjer fra den nordiske MDS-gruppen, Nordic Care Programme, sist oppdatert i desember 2011.
Risiko for transformasjon til AML og forventet overlevelse kan estimeres ut i fra det internasjonale prognostiske scoringssystemet for MDS, IPSS for MDS [3]. Poengsystemet muliggjør å dele pasientene inn i fire grupper: Lavrisk, intermediær-1, intermediær-2 og høyrisk. Poengene settes på bakgrunn av informasjon som er lett tilgjengelig med standard prøvetaking. Antall cytopenier bestemmes enkelt ut i fra hematologiske blodprøver med differensialtelling. Hb under 10 g/dL, nøytrofile under 1,8 x 10*9/L og platetall under 100 x 10*9/L defineres som cytopenier. Deretter må man telle antall blaster i benmargen og sende benmarg til kromosomundersøkelse (cytogenetikk). For nøyere klassifisering av hver undergruppe av MDS har man WHO-klassifikasjonen av hematopoietiske og lymfoide neoplasier fra 2008.
 Som en tommelfingerregel kan man tilnærme seg en pasient der man mistenker MDS ved først og fremst ekskludere andre årsaker til cytopeni og dysplasi, deriblant B-vitaminmangler, virale infeksjoner som HIV og hepatitter, medikamentutløst cytopeni og cytotoxisk benmargssakade, anemi ved kronisk sykdom, autoantistoff, kronisk leversykdom, alkoholmisbruk, eksponering for tungmetaller som kvikksølv og bly, aplastisk anemi, paroxysmal nokturnal hematuri, myelofibrose og akutt myelogen leukemi.
Som en tommelfingerregel kan man tilnærme seg en pasient der man mistenker MDS ved først og fremst ekskludere andre årsaker til cytopeni og dysplasi, deriblant B-vitaminmangler, virale infeksjoner som HIV og hepatitter, medikamentutløst cytopeni og cytotoxisk benmargssakade, anemi ved kronisk sykdom, autoantistoff, kronisk leversykdom, alkoholmisbruk, eksponering for tungmetaller som kvikksølv og bly, aplastisk anemi, paroxysmal nokturnal hematuri, myelofibrose og akutt myelogen leukemi.
For prøvetaking til benmargsutstryk er det tilstrekkelig å aspirere et par dråper benmarg fra sternum. Benmargen skal farges med May-Grünewald-Giemsa-farging og jernfarging. Ved utvidet prøvetaking til immunfenotyping og cytogenetikk bør man aspirere fra crista iliaca. Man trekker da opp 2-3 ml benmarg til hver undersøkelse i en heparinisert sprøyte. Det er viktig å vende på sprøyten for å forhindre koagulasjon. Prøven til cytogenetikk overføres sterilt til et eget rør med transportmedium og forsendes etter lokale prosedyrer til nærmeste laboratorium som utfører analysen. Det er viktig å unngå at prøven blir liggende over helg. Man bør verifisere den morfologiske diagnosen etter to måneder ved å ta cristabiopsi. Hos voksne gjøres benmargspunksjon i lokalanestesi, men hos barn er det nødvendig med full narkose og er en spesialistoppgave for pediatere.
Behandling av MDS bestemmes på bakgrunn av pasientens alder og risikoprofil. Cytogenetikk er særdeles viktig da denne er vist å være en robust prognostisk variabel uavhengig av terapivalg. Analyser av genuttrykk er foreløpig lite anvendt, da de kjente genetiske markørene er relativt sjeldne, uspesifikke og ikke vist å være prediktivt eller prognostisk signifikante. I tillegg er metodene for genanalyser ved MDS lite standardisert mellom de ulike sentrene. Imidlertid begynner man nå å nærme seg standardisering av metoder for flowcytometriske analyser av benmarg fra pasienter med MDS. Undersøkelsen går ut på at spesifikke fargemerkede antistoff bindes til ulike proteiner eller markører som allerede finnes på cellene. Cellene sendes deretter i en væskestrøm gjennom et apparat som lyser på cellene med lasere og måler størrelse og granularitet, samt registrerer fargesignaler fra de cellene der antistoffene har bundet seg. Man kan her se spesifikke kvalitative forskjeller som gjenspeiler funnene man ser i vanlig lysmikroskopi av benmarg, for eksempel hypogranulering av nøytrofile granulocytter. I tillegg kan man ved hjelp av immunologiske markører tydeliggjøre unormal utmodning av cellene og vise aberrante uttrykk av myeloide overflatemarkører, samt å påvise uttrykk av ikke-myeloide antigen [4-6].
Hos alle pasienter under 60-70 år med MDS som ikke befinner seg i den laveste risikogruppen, må allogen stamcelletransplantasjon vurderes. Det er vist at yngre pasienter med hypoplastisk MDS og en spesiell fenotype kan ha nytte av immunterapi. For behandlingsvalg vises det til de nevnte handlingsprogrammer, hvor man vil finne detaljerte beskrivelser av fremgangsmåte for vekstfaktorbehandling, chelerende eller hypometylerende behandling. Ved svært høyt blastantall vil man også kunne velge å gi cellegiftregimer tilsvarende de som brukes ved AML.
Det finnes flere hypoteser på hvordan en myelodysplastisk klon oppstår. Man tenker seg at sykdommen oppstår på stadiet ”committed myeloid precursor” i utviklingen av de hematopoietiske stamcellene. Hos lavrisikopasientene er immunsystemet ofte svært aktivert med et høyt nivå av inflammatoriske cytokiner i benmargen. Disse cytokinene virker myelosupprimerende. I tillegg forekommer det kloner av cytotoksiske T-celler som antas å angripe progenitorcellene. Hematopoiesen blir ineffektiv og det sees en stor andel apoptotiske celler. Etter hvert som sykdommen utvikler seg, forsvinner tendensen til apoptose, og den maligne klonen dominerer benmargen, med en stigende andel umodne myeloide forstadier. Man ser også at den immunologiske responsen forandrer seg til å bli immunsupprimerende i stedet for pro-inflammatorisk. Selv om det finnes flere bevis på et dysregulert immunsystem ved MDS, har vi ingen kunnskap om forandringene oppstår som et svar på den maligne klonen eller om den maligne klonen fremdyrkes av de immunologiske forandringene.
MDS er en heterogen gruppe stamcellesykdommer. Moderne diagnostikk er basert på blodprøver, benmargsmorfologi og cytogenetikk og på bakgrunn av disse undersøkelsene kan man dele pasientene inn i ulike risikogrupper. Mengde blaster i benmargen (under 20 prosent) er kritisk for å utelukke AML. Kvalitative undersøkelser med væskestrømcytometri vil trolig snart få diagnostisk betydning. Noen få pasienter vil kunne tilbys potensielt kurativ behandling med allogen stamcelletransplantasjon, men for den resterende pasientmassen vil behandlingen være palliativ og symptomrettet.



