
Frank Brosstad.
Av Frank Brosstad, Professor, Institutt for indremedisinsk forskning, OUS Rikshospitalet
Blodårer er innvendig kledd med et encellet lag av endotelceller. Ved karveggskade vil endotelcellelaget skades og blodplater kommer i kontakt med et kompleks av von Willebrand faktor (vWF) og kollagen i det subendoteliale rom.
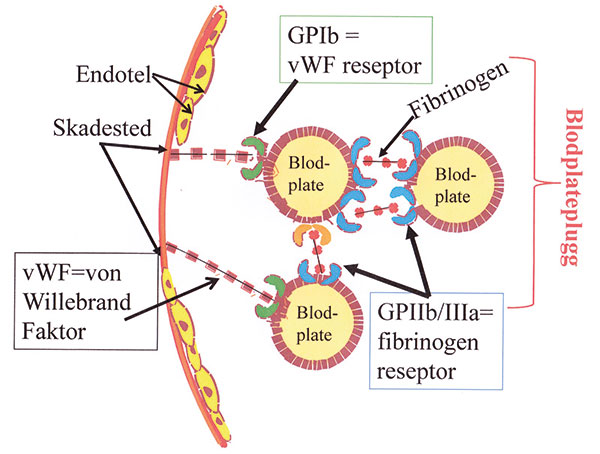
Figur 1, Figuren fremstiller: 1) blodplateadhesjon ved binding av von Willebrand Faktor (vWF) i subendotelet til blodplatens vWF-reseptor. 2) blodplateaggregasjon og blodplatepluggdannelse via binding av fibrinogen til blodplatenes fibrinogen reseptorer.
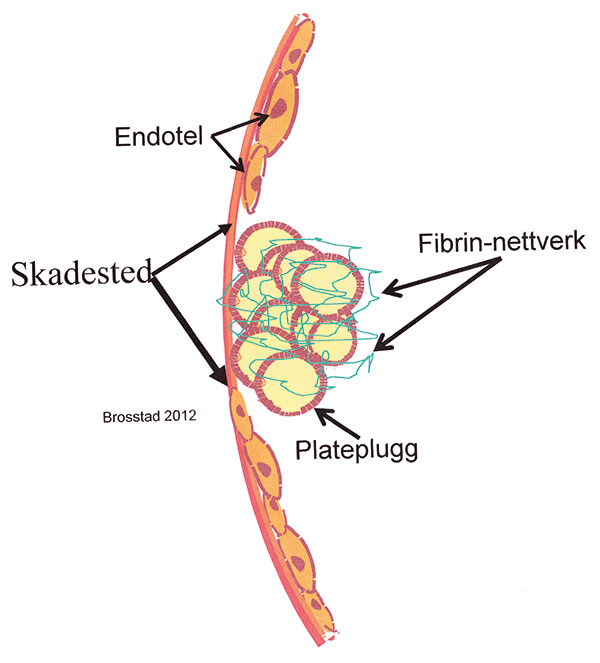
Figur 2. Figuren fremstiller koagulasjonsfaktoraktiveringen på blodplatepluggens overflate med dannelse av fibrin som forsterker platepluggen mekanisk.
Det er dette fenomenet vi kaller fysiologisk hemostase [1]. Men en plateplugg kan også dannes på overflaten av et krakelert/rumpert ateromatøst plakk i en arterievegg ved at blodplatene kommer i kontakt med plakkets innhold av blodplateaktiverende materiale (kollagen, vevsfaktor, trombin) (Figur 3).
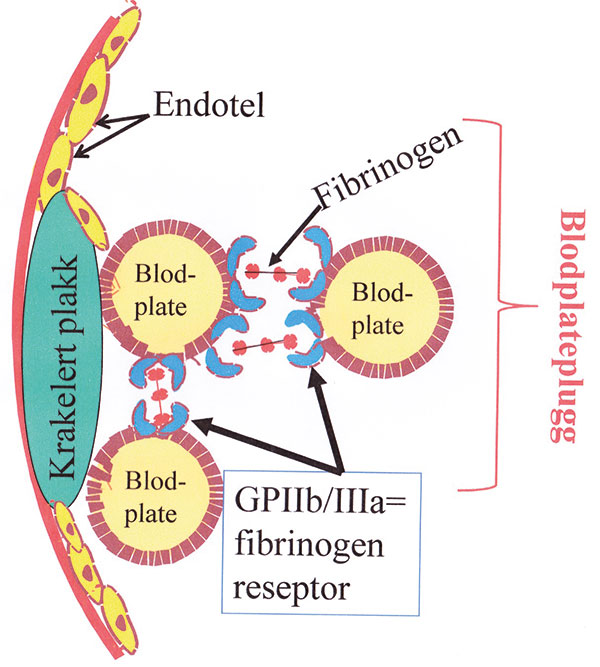
Figur 3. Figuren fremstiller platepluggdannelse på overflaten av et krakelert plakk i en arterie, som kan forårsake aterotrombose.
Siden hjertets og hjernens arterier er endearterier (liten eller ingen kollateral blodsirkulasjon til arteriens forsyningsområde), kan resultatet bli nedsatt eller opphevet sirkulasjon med infarsering (cerebrokardial aterotrombose) (Figur 4).
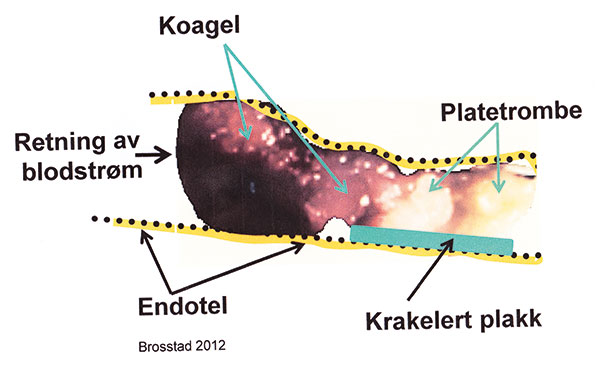
Figur 4. Figuren fremstiller aterotrombose i en koronararterie. Legg merke til trombens «hvite hode» (= plateplugg/platetrombe) og dens «røde kropp» som skyldes koageldannelse hvor alle blodets elementer er til stede.
Blodplaten har altså en sentral rolle i utviklingen av cerebrokardial aterotrombose, og inhibisjon av blodplatens adhesjons- og aggregasjonsevne kan slik gi god anti-aterotrombotisk profylakse.
Kunnskapen om blodplatens aktiverings- og inaktiveringsmekanismer har derfor resultert i platehemmende medikamenter, som har vist seg å være effektive i kliniske studier.
1) aktivering av platens fosfolipase A2 som frigjør arakidonsyre fra platemembranens innside. Arakidonsyre omsettes av platens cyklooksygenase-1 (COX 1) via intermediater til tromboksan A2 (TxA2). TxA2 skilles ut i platens nærmiljø og aktiverer naboplater ved binding til og stimulering av deres TxA2 reseptorer [2]. Dermed genereres et transmembrant signal som aktiverer platenes fibrinogenreseptorer (GPIIb/IIIa, ca. 50 000 per plate). Fibrinogen kan dermed binde aktiverte plater sammen og danne en plateplugg (fysiologisk hemostase, Figur 1-2) eller en platetrombe (patofysiologisk aterotrombose Figur 3- 4). Dannelse av plateplugg og platetrombe har således et felles biokjemisk og cellefysiologisk grunnlag.
Acetylsalisylsyre inaktiverer platenes cyklooksygenase-1 og dermed platenes TxA2 produksjon, en effekt som varer hele platens levetid (7-10 dager). Antallet av pasienter som mangler denne effekten av acetylsalisylsyre (ASA resistens), er omdiskutert, men antagelig lite. (Figur 5) [2].
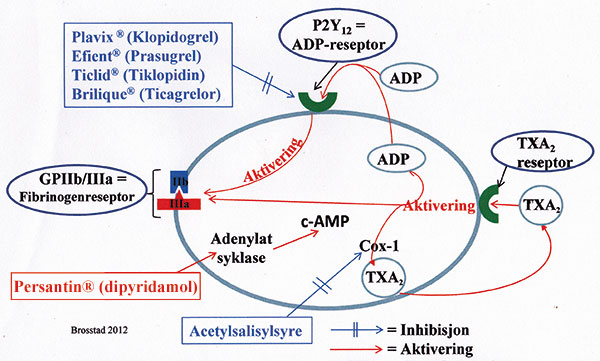
Figur 5. Aktivering av blodplatens fibrinogenreseptor er en forutsetning for blodplatepluggdannelse og for aterotrombose. Figuren viser hvordan dette kan skje via agonistene TXA2 og ADP, og hvilke medikamenter som hemmer aktiveringen; 1) ved stimulering av adenylat syklase som øker platens c-AMP (Persantin® (Dipyridamol)), 2) ved enzyminaktivering av Cox-1 (acetylsalisylsyre) eller 3) ved blokkering av ADP reseptor P2Y12 (Plavix,Ticlid, Efient, Brilique).
2) frigjøring av ADP fra platens granula skilles ut og binder seg til platens og naboplaters ADP reseptor (P2Y12) (Figur 5). Binding til platens ADP-reseptorer genererer transmembrane signaler som aktiverer fibrinogenreseptor, og dermed dannelse av plateplugg/platetrombe. Medikamentell hemming av ADP-indusert plateaggregasjon har vært mulig i de 20 siste årene [3] ved bruk av ticlopidin og klopidogrel, men nå har også nye medikamenter kommet.
Resultatene av studier der effekten av klopidogrel har vært sammenlignet med prasugrel og ticagrelor har ført til at European Society of Cardiology anbefaler behandling av UAP/NSTEMI med de to sistnevnte fremfor klopidogrel [6]. Dette kan også få snarlig betydning for vurderingen av gjeldende retningslinjer for behandlingen av STEMI.
Dipyridamol (Persantin®, Asasantin Retard®)[7] stimulerer adenylatsyklase og hemmer fosfodiesterase i platen slik at konsentrasjonen av platens c-AMP nivå øker. Derved nedsettes platens reaktivitet. Dipyridamol øker ikke blødningstendens i kombinasjon med acetylsalisylsyre eller warfarin. Midlet brukes ofte kombinert med acetylsalisylsyre (Asasantin Retard®) i hjerneslagprofylakse[7]. Halveringstiden er cirka 3 timer med utskillelse via lever. (Figur 5)
Effektive medikamenter med evne til å hemme blodplatens aktiveringssignaler og dens aterotrombotiske potensial, gjennom enzyminduksjon (dipyridamol), enzyminaktivering (acetylsalisylsyre) eller reseptorblokade (klopidogrel, prasurgrel, ticagrelor), står til disposisjon. Prisen ved bruk er en økt klinisk blødningstendens, som i de fleste tilfeller vil oppveies av en desto bedre antitrombotisk effekt. Midlenes farmakodynamikk i interaksjon med andre medikamenter krever imidlertid oppmerksomhet, kunnskap og erfaring av så vel terapeut som pasient. I nær fremtid vil det komme nye platehemmere som påvirker andre av blodplatens aktiveringsmekanismer og signalveier, og som vil gi nye terapeutiske muligheter og utfordringer.



