
Anne Naalsund
Anne Naalsund, fhv overlege, Lungeavdelingen, OUS Rikshospitalet. Trond Mogens Aaløkken, Overlege, Radiologisk avdeling, OUS Rikshospitalet og Tone Sjåheim, Overlege, Lungeavdelingen, OUS Rikshospitalet.
De siste tiår har kunnskapen økt vedrørende klassifikasjon, utvikling og behandling av idiopatiske intersitielle pneumonier. Idiopatisk interstitiell fibrose forekommer hyppigst. Artikkelen gir en oversikt over alle de syv undergruppene.
Idiopatiske interstitielle pneumonier (IIP) er en gruppe diffuse parenkymatøse lungesykdommer av ukjent etiologi karakterisert ved ulike grader av inflammasjon og fibrose, og tilstrekkelig ulike til å oppfattes som separate sykdommer. I henhold til American Thoracic Society/ European Respiratory Society (ATS/ERS) consensus klassifikasjon i 2002 (1) omfatter IIP syv tilstander, hvorav den hyppigst forekommende, idiopatisk pulmonal fibrose (IPF) utgjør ca 50 % (Figur 1). Klassifikasjonen ble utarbeidet ved ”expert opinions” og ”consensus” innen forfattergruppen heller enn tuftet på evidensbasert kunnskap, men representerte likevel et gjennombrudd internasjonalt fordi det for første gang ble oppnådd enighet om en felles klinisk, radiologisk og patologisk klassifikasjon av de sju sykdomsbilder som utgjør IIP. Dokumentet har hatt betydning for klinisk håndtering av pasienter med mistenkt IIP, har fungert som basis for studiedesign for kliniske studier og stimulert til klinisk forskning på områder der faktisk kunnskap har vært mangelfull. Med utgangspunkt i dette dokumentet ble det i 2011 publisert evidensbaserte internasjonale retningslinjer for diagnostikk og oppfølging av IPF alene (2). På grunn av ny kunnskap særlig når det gjelder medikamentell behandling, er disse allerede gjenstand for revisjon, nå sammen med de øvrige IIP´er.

Fig. 1: Oversikt i henhold til American Thoracic Society/ European Respiratory Society guidelines 2002.
I diagnostikk av IIP står HR-CT sentralt, særlig når det morfologisk gjelder å skille idiopatisk pulmonal fibrose fra øvrige IIP, men også når det gjelder å skille mellom de øvrige seks.
En nærmere omtale av de syv idiopatiske sykdommene følger nedenfor, med særlig vektlegging på idiopatisk pulmonal fibrose (IPF) som utgjør mer enn 50 % av IIPer.
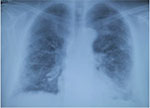
Fig. 2A: Viser fingerclubbing. Mann f.1937 med idiopatisk pulmonal fibrose. Røntgen thorax viser fortetninger mest i basale avsnitt. HRCT viser perifere fortetninger med bikakemønster og traksjonsbronkiektasier.
Idiopatisk pulmonal fibrose (IPF) er definert som en spesifikk form for kronisk progredierende interstitiell pneumoni av ukjent årsak begrenset til lungen (3).
– Histopatologisk foreligger et såkalt ”usual interstitial pneumonia mønster” (UIP-mønster):
– Uttalt fibrose/arkitektonisk destruksjon av lungevev med utvikling av bikakemønster subpleuralt /paraseptalt. Bikakemønstret skyldes parenkymdestruksjon resulterende i perifere cyster med vegger av kollagen kledd av cilieførende epitel.
– Heterogenisitet i utbredelse av forandringer, og i alder av fibrosen.
– Påvisning av flekkvise fibroblastfoci.
Diagnosen krever eksklusjon av kjente årsaker til UIP, som yrkeseksponering, særlig asbest, medikamentskade eller kjent systemsykdom. Avanserte former for sarkoidose og kronisk hypersensitivitetspneumonier kan også være viktige imitatorer og må elimineres.
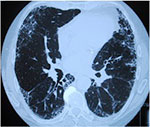
Fig. 2B: Viser fingerclubbing. Mann f.1937 med idiopatisk pulmonal fibrose. Røntgen thorax viser fortetninger mest i basale avsnitt. HRCT viser perifere fortetninger med bikakemønster og traksjonsbronkiektasier.
Mens det er vanlig å anta at fibrose er enderesultatet av ulike former for inflammasjon, sees ved IPF svært beskjeden grad av slike forandringer. Fibrosen ser ut til å oppstå ”de novo” som et resultat av repeterende diffust utbredte epitelskader i alveolepitelet over tid, muligens en forklaring på hvorfor antiinflammatoriske medikamenter som kortikosteroider ikke har effekt ved denne tilstanden (4-6). Graden av destruksjon er nemlig så uttalt at selv om framtidige behandlingsformer ville kunne reversere milde grader av fibrose, er det utenkelig at lungens struktur og/eller funksjon vil kunne gjenopprettes i disse tilfellene. Behandlingsmål ved utprøving av medikamenter har derfor endret seg fra et ønske om å reversere sykdommen til å kunne forsinke eller hindre utvikling og progresjon av kronisk fibrose.
Prevalens- og insidenstallene er usikre både pga endringer i terminologi og inntil nylig også på grunn av mangel på diagnostiske kriterier. En norsk studie beregnet prevalens av IPF til 23,4/100.000 og insidens 4,3/100.000 (7). I Europa ellers er insidenstall rapportert mellom 0,22 og 7,4/100.000, og forekomsten øker (8).
Oftest rammer sykdommen mannlige røykere eller eks-røykere over 50 år, og prevalens og insidens øker med alderen. Forløpet varierer, men sykdommen er forbundet med svært dårlig prognose – median overlevelse er 2 – 5 år, altså verre enn for mange cancersykdommer (9).
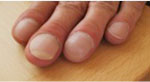
Fig. 2C: Viser fingerclubbing. Mann f.1937 med idiopatisk pulmonal fibrose. Røntgen thorax viser fortetninger mest i basale avsnitt. HRCT viser perifere fortetninger med bikakemønster og traksjonsbronkiektasier.
Dominerende symptomer er økende dyspne, ofte også tørrhoste. Mer enn halvparten av pasientene har fingerclubbing (Figur 2). De fleste utvikler respirasjonssvikt, mange også ulike grader av pulmonal hypertensjon. Ca 10 % av pasientene har et forløp preget av akutte forverrelser av dyspne som utvikles i løpet av dager/ uker – en situasjon forbundet med høy mortalitet (3, 10). I denne situasjonen gis fortsatt kortikosteroider selv om behandlingseffekten ikke er dokumentert.
Ved autopsi sees forandringer forenlig med IPF komplisert av akutt interstitiell pneumoni i form av diffus alveolær skade (Kfr punkt 4).
HR-CT står sentralt når det gjelder å stille diagnosen IPF (Figur 2). De karakteristiske morfologiske forandringene er påvisning av subpleurale retikulære fortetninger mest uttalt basalt. Bikakemønster med cyster med diameter 2-20 mm sees hos > 90 % av pasientene på diagnosetidspunktet. Traksjonsbronkiektasier er et hyppig funn. Forutsatt at disse forandringene er til stede, er de så nært korrelert til histopatologiske funn (90 – 100%) at kirurgisk biopsitaking ikke er indisert (11, 12) . Dette gjelder ca 2/3 av pasienter med IPF.
Det anbefales at diagnostikk av IPF skjer i et samarbeid mellom kliniker, radiolog og patolog (1, 3). Et slikt samarbeid har redusert behovet for kirurgisk lungebiopsi.
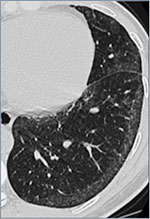
Fig. 3A: Non-spesifikk
interstitiell pneumoni.
Cellulær variant. Perifere fortetninger med mattglasspreg.
Behandling av IPF er utfordrende, og økende forekomst av sykdommen sammenholdt med svært dårlig prognose har ført til forenet innsats fra medisinsk forskning og farmasøytisk industri for å finne fram til effektiv terapi. De aller fleste publiserte studier har vist negative resultater, men har også ført til registrering av det første medikament med dokumentert effekt når det gjelder å forsinke fall i lungefunksjonen. Pirfenidon (Esbriet) er et medikament med visse antifibrotiske, antiinflammatoriske og antioxidative egenskaper som i flere kontrollerte studier har vist seg å ha en forsinkende effekt på utvikling av fibrose hos pasienter med mild moderat form for IPF (13-15). Medikamentet er registrert i Norge.
I retningslinjene av 2011 (3) er bare to behandlingsmodaliteter anbefalt med en viss tyngde: Lang-tids oksygenbehandling til pasienter med hypoksemi og lungetransplantasjon dersom pasienten ellers er egnet for slik behandling. Lungetransplantasjon er den eneste behandlingsform som har vist bedret overlevelse. I England representerer pasienter med IPF 20 % av alle lungetransplantasjoner. Likevel utgjør de majoriteten av pasienter som dør på venteliste.
For pasienter < 60 år, er henvisning til vurdering/ utredning for lungetransplantasjon på Rikshospitalet aktuelt. I Norge har ca 50 pasienter med IPF eller andre former for IIP gjennomgått transplantasjon.
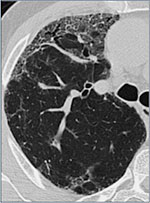
Fig. 3B: Non-spesifikk interstitiell pneumoni.
Fibrotisk variant. Perifere retikulære fortetninger med bikakemønster, lite mattglassforandringer.
Lindrende behandling rettet mot plagsomme symptomer (hoste, dyspne) og mot sykdomsspesifikke komplikasjoner som pulmonal hypertensjon, infeksjoner og mot gastroøsofageal refluks (GERD) står ellers sentralt. Mange studier har vist økt forekomst av GERD hos pasienter med IPF, og samtidig har bare ca 50% av disse symptomer fra sin reflukssykdom. Det er holdepunkter for at kronisk trakeobronkial aspirasjon over tid kan bidra til progresjon av IPF, sannsynligvis også øke antallet episoder med akutte forverrelser. Pasienter med IPF bør derfor utredes mhp refluks, og alle med påvist reflukssykdom behandles – uavhengig av om de har symptomer eller ikke (3).
I ATS/ERS consensus dokumentet av 2002 ble Non-spesifikk interstitiell pneumoni (NSIP) vurdert å være en midlertidig diagnose for ikke-klassifiserbare idiopatiske pneumonier, muligens en variant av IPF (1). Etter en nyere gjennomgang i regi av American Thoracic Society er det nå besluttet at idiopatisk NSIP er en spesifikk klinisk patologisk diagnose med relativt god prognose: 5 og 10 års overlevelse henholdsvis 80% og 73% (16). Tilstanden affiserer oftest pasienter i 40 – 50 års alder, kvinner noe hyppigere enn menn. Symptomene er kronisk progredierende dyspne og tørrhoste, mens clubbing forekommer langt sjeldnere enn hos pasienter med IPF.
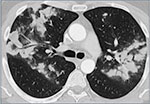
Fig. 4A: Kryptogen organiserende pneumoni. Områder med konsolidering i begge lunger, perifer dominans.
Histologisk sees varierende grader av inflammasjon (cellulær variant) og fibrose (fibrotisk variant) – en inndeling som kan være komplisert, men som ser ut til å ha prognostiske implikasjoner (17–19). Fibrose ved NSIP er homogent utbredt, og patologisk er fibroblastfoci og bikake-mønster sjeldne. Som ved IPF kan et identisk histologisk UIP-bilde ses hos pasienter med systemiske bindevevssykdommer, medikamentelt utløst lungesykdom og ved enkelte tilfeller av hypersensitivitetspneumonitt (20-21). Disse tilstandene må elimineres før diagnosen NSIP stilles.
Radiologisk er bilaterale og symmetriske mattglassfortetninger et sentralt funn, oftest med perifer og basal utbredelse (22) (Figur 3). Sparing av det subpleurale rom forekommer. Fine subpleurale retikulære fortetninger, traksjonsbronkiektasier og skrumpning med strukturelle forandringer reflekterer fibrose. Først og fremst er det påvisning av mattglassfortetninger og fravær av dominerende bikakemønster som skiller NSIP fra IPF.
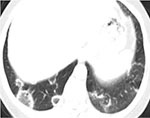
Fig. 4B: Kryptogen organiserende pneumoni. Omvendt halotegn, også kalt Atolltegn.
Særlig pasienter med den cellulære varianten responderer godt på behandling med kortikosteroider.
Kryptogen organiserende pneumoni (COP), tidligere kjent som bronchiolitis oblitterans organiserende pneumoni (BOOP), affiserer primært alveoler, ducti alveoli og små luftveier, undertiden også interstitiet. Fibrose er sjelden. Det histologiske bildet er dominert av granulasjonsvev/ polypper som også kan sees ved ulike tilstander som infeksjoner, bindevevssykdommer og medikamentskade. Ved funn av organiserende pneumoni i biopsier, må derfor tilgrunnliggende årsak til sykdommen elimineres.
Klinisk debuterer tilstanden med raskt innsettende influensalignende symptomer, hoste og åndenød, og
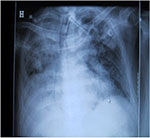
Fig. 5A: Akutt interstitiell pneumoni. Rtg thorax med utbredte lungefortetninger hos intubert pasient.
imiterer og forveksles dermed ofte med infeksjon (23, 24).
Radiologisk sees bilaterale flekkvise konsoliderte fortetninger med subpleural eller peribronkial dominans, og med eller uten mattglassfortetninger. Dessuten kan rundaktige fortetninger med mattglass og sentral oppklaring, det såkalte Atolltegn eller omvendt halotegn sees hos et fåtall av pasientene (Figur 4). Dette er sagt å være et nærmest patognomonisk funn (25).
COP responderer svært godt på steroider i mer enn 80% av tilfellene. Tilbakefall skjer ofte, i regelen fordi behandlingen har vart for kort (26). Undertiden må pasientene behandles mer enn 12 måneder på tross av rask klinisk og radiologisk respons initialt. Også tilbakefall kan behandles med kortikosteroider. Ved mangelfull respons kan det være indisert å forsøke steroidsparende medikasjon som cyklofosfamid eller azathioprin.
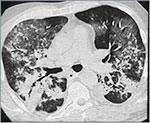
Fig. 5B: CT: Massive bilaterale lungefortetninger, blandet bilde av konsolidering og mattglass.
Akutt interstitiell pneumoni (AIP), også kalt Hamman-Rich syndrom, er en sjelden fulminant form for idiopatisk akutt lungeskade karakterisert ved akutt debut og rask progresjon til respirasjonssvikt. Feber sees ofte. Det er ingen kjønnsdominans, og gjennomsnittsalder ved sykdomsdebut er 50 – 55 år. Røyking er ingen risikofaktor.
Ved biopsi sees diffus alveolskade, identisk med histologiske forandringer og stadier ved ”adult respiratory distress syndrome” (27).
Differensialdiagnostisk må Pneumocystis jirovecii, kardial stuvning og akutt forverring av IPF overveies.
Radiologisk sees bilaterale flekkvise mattglassfortetninger og også områder med konsolidert lungevev (Figur 5). Arkitektonisk destruksjon av lungeparenkymet pga fibrose samt traksjonsbronkiektasier utvikles raskt. Det er god korrelasjon mellom histopatologiske stadier av AIP og de radiologiske forandringer (28).
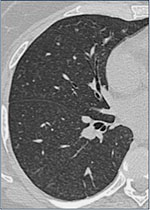
Fig. 6A: Respiratorisk bronkiolitt assosiert med interstitiell lungesykdom. Tettliggende centrilobulære nodulære fortetninger.
Ingen medikamentell behandling har vist seg å ha effekt, mekanisk støttebehandling med respirator er ofte påkrevet. De fleste pasienter med AIP får kortikosteroider og antibiotika på empirisk basis. Det finnes ingen overbevisende dokumentasjon for effekt av slik behandling. Korttidsprognosen er svært dårlig med over 60 % mortalitet i løpet av de første 6 måneder.
Disse to tilstandene har flere fellestrekk klinisk og patologisk, og mange mener at deskvamativ interstitiell pneumoni (DIP) representerer en videreutvikling av respiratorisk bronkiolitt interstitiell pneumoni (RB-ILD) (29, 30). Begge tilstander er assosiert med eksponering for tobakksrøyk, og begge er karakterisert ved opphopning av pigmentmakrofager, peribronkiolært ved RB-ILD og mer generelt utbredt ved DIP.
Kronisk dyspne og tørrhoste av varierende grad er dominerende symptomer. Sykdommene affiserer røykere i 40 – 50 års alder, ingen kjønnsdominans.
På HR-CT sees ved RB-ILD fortykkelse av bronkialvegger, centrilobulære noduli og områder med
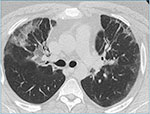
Fig 6B: Deskvamativ interstitiell pneumoni. Bilaterale mattglassfortetninger.
luftretensjon (”air-trapping”) (Figur 6). Det er ingen lobær predominans. Mattglassfortetninger er mer uttalt ved DIP, og da oftest uttalt i deklive avsnitt. Små cyster kan også sees.
Behandling er primært røykestopp – hvis vellykket, er det et tiltak som har vist seg å kunne føre til remisjon, først og fremst av RB-ILD. Ved mer utbredt sykdom gis kortikosteroider – primært ved DIP, og da oftest med godt behandlingsresultat. Tilbakefall sees – ofte ved gjenoppstart av røyking (31). Prognosen er god, selv om enkeltpasienter med DIP kan utvikle mer generalisert lungefibrose og respirasjonssvikt.
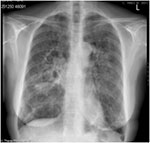
Fig 7A: Lymfocyttær interstitiell pneumoni.
Rtg thorax med bilaterale fortetninger og enkelte cyster.
De fleste pasienter med histopatologisk lymfocyttisk interstitiell pneumoni (LIP) er assosiert med Sjøgren´s syndrom. Inkludering av LIP i gruppen IIP er kontroversiell, men gjøres fordi enkeltpasienter har vist seg å ha en idiopatisk variant. LIP kan klinisk og radiologisk gi mistanke om malign sykdom, og dette må utelukkes.
Histopatologisk er LIP en ekte interstitiell lungesykdom med fortykket interstitium forårsaket av tette lymfoide infiltrater, men ellers med beskjedne forandringer peribronkialt og i alveolene.
Klinisk debut er uspesifikk, ofte med kronisk progredierende hoste og dyspne. Enkelte har i tillegg feber, artralgier og vekttap. Kvinner affiseres oftere enn menn, de fleste i 50-års alder. På tross av at spirometriverdier i regelen bare er lett redusert, har de fleste pasienter betydelige gassvekslingsforstyrrelser. Bronkoalveolær lavage (BAL) kan vise lett lymfocytose.
Radiologisk sees diffuse mattglassfortetninger samt centrilobulære noduli, fortykkede bronkovaskulære strukturer, forstørrede lymfeknuter i hilus og mediastinum, og oftest tynnveggede cyster i lungeparen-kymet (Figur 7). Kombinasjonen mikronoduli, cyster og utbredte mattglassforandringer er suggestivt for denne diagnosen (32).
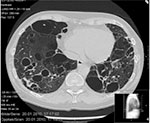
7B: CT: Utbredte cystiske forandringer. Samme pasient som A etter behandling med kortikosteroider der fortetninger er gått tilbake mens cystene persisterer.
Det foreligger ingen gode data når det gjelder behandlingseffekt, oftest anvendes kortikosteroider eventuelt med tilskudd av cytostatika – sendoxan eller azathioprin. Sykdommen kan undergå malign transformasjon. Enkelte pasienter utvikler diffus lungefibrose med respirasjonssvikt og 5-års overlevelse er ca 60 %.
IIP omfatter en undergruppe av interstitielle lungesykdommer – eller mer korrekt: diffuse parenkymatøse lungesykdommer – som har vært mye omtalt i medisinsk litteratur de siste 10 – 12 år. Det er sannsynlig at nomenklatur, klassifikasjon og behandling av IIP´er vil endres i årene som kommer, i takt med ny kunnskap om patofysiologi ved de enkelte tilstandene, og etter hvert som resultater fra større kontrollerte multisenter studier foreligger.
HR-CT har en viktig rolle i diagnostikk av IIP ettersom det radiologiske bildet ved disse sykdommene er godt beskrevet, og i mange tilfeller godt korrelert til patologisk morfologi. Særlig gjelder dette IPF. Det sees imidlertid også en betydelig grad av overlapping mellom disse funnene, og det nødvendiggjør et tett samarbeid mellom kliniker, radiolog og patolog for å sikre korrekt diagnose.



