
Rannveig Skrunes.
Av Rannveig Skrunes, konstituert overlege, medisinsk avdeling, Haukeland Universitetssykehus og Einar Svarstad, professor og seksjonsoverlege, Universitetet i Bergen og medisinsk avdeling, Haukeland Universitetssykehus.

Einar Svarstad.
Med økende kunnskap om genomet følger ny innsikt i årsak til enkelte nyresykdommer. Flere nye gener er identifisert som medvirkende i utvikling av nyresykdom, og den genetiske årsaken til kjente kliniske entiteter er i enkelte tilfeller også avslørt. Gode helseregistre, som for eksempel Norsk Nefrologiregister, gjør det mulig å undersøke eventuell opphopning av nyresykdommer i den norske populasjonen. Uromodulin-assosiert nyresykdom og Fabry sykdom omtales som eksempler på «nye» sykdommer med variable fenotyper som indremedisinere bør ha kjennskap til.
Forekomsten av kronisk nyresykdom har vært økende verden over (1, 2), men insidensen av endestadie nyresvikt (ESRD) varierer fra land til land, og mellom forskjellige etniske grupper. Asiater i USA har for eksempel en mye lavere insidens enn både afro-amerikanere og amerikanere av europeisk herkomst (3). Selv innad i land med en relativt homogen befolkning, som Norge, kan man se forskjeller i insidens av ESRD (4). Vi kjenner ikke årsaken til hvorfor for eksempel rogalendingene tilsynelatende har bedre nyrehelse enn hedemarkingene (4). Slike forskjeller kan ha mange forklaringer.
Den kliniske hverdagen kan gi inntrykk av at noen familier er overrepresenterte i nyrepoliklinikken, i dialysepopulasjonen og i transplantasjonspopulasjonen. Mange av disse har allerede kjente hereditære nefropatier, men systematisk undersøkelse av registerdata og helseinformasjon fra pasienter som mottar nyreerstattende behandling, åpner for å undersøke muligheten for at også andre typer nyresvikt akkumuleres i familier.
Adult polycystisk nyresykdom (APKD) er den vanligste arvelige nyresykdom og utgjør ca 10 % av alle med ESRD både i Norge og resten av verden (4,5). Nye terapimuligheter som forsinker cysteutvikling og således nyresvikt, er under rask utvikling og vil i løpet av få år trolig være i vanlig bruk.
Fremskritt i kartlegging av arvemateriale har de senere år medvirket til at en rekke nye sykdomsassosierte mutasjoner er påvist. Begrepet «sjeldne sykdommer» har fått et nytt innhold når omfanget av slike tilstander nærmer seg 10 % av enkelte vestlige befolkningsgrupper. To eksempler på relativt nylig påviste mutasjoner, relevante for diagnostikk og behandling i nefrologisk og indremedisinsk praksis, skal omtales kort nedenfor, Det er mutasjoner i uromodulin-genet (UMOD), som ofte er assosiert med tidlig forekomst av urinsyregikt (6), og mutasjoner i alfa-galaktosidasegenet (GLA) som er årsak til Fabry sykdom, en systemisk sykdom med manifestasjoner fra en rekke organsystemer (7).
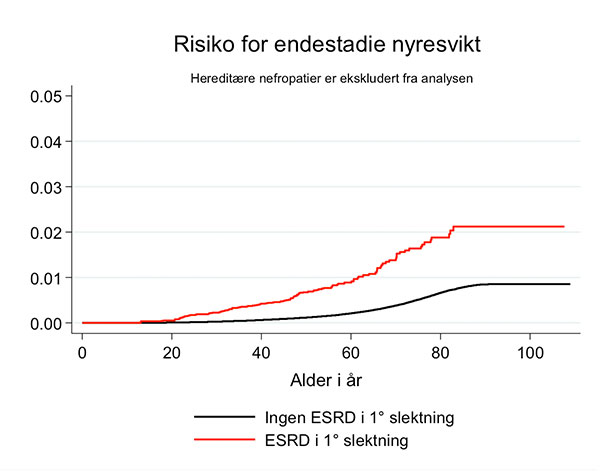
Figur 1. Risiko for end-stage renal disease (ESRD) i henhold til om det er identifisert en førstegradsslektning med ESRD eller ei.
Norsk Nefrologiregister ble etablert i 1980, og mottar årlig klinisk informasjon om pasienter som får nyreerstattende behandling (dialyse eller transplantasjon). Registeret gjør det mulig å følge en enkelt pasient i et behandlingsforløp, men det gjør det også mulig å finne familier med opphopning av ESRD. Dette kan gi mulighet for tidlig diagnostikk og intervensjon hos familiemedlemmer med arvelig nyresykdom.
For å se nærmere på denne problemstillingen ble data fra Norsk Nefrologiregister koblet opp mot Folkeregisteret og Dødsårsaksregisteret. Disse koblingene gjorde det mulig å strukturere informasjon slik at en familiestreng med indeksperson og førstegradsslektninger av indekspersonen kunne konstrueres. Diagnosekoder for alle som mottok, eller hadde mottatt, nyreerstattende behandling, ble hentet fra Norsk Nefrologiregister, dødsår ble hentet fra Dødsårsaksregisteret der det var aktuelt.
Analyse av disse dataene viser at det også i den norske populasjonen er slik at nyresykdommer akkumuleres i familier. Risikoen for å utvikle ESRD selv er naturlig nok størst dersom det allerede foreligger en kjent hereditær nefropati i familien (Figur 1). Relativ risiko for ESRD i den norske populasjonen, dersom en førstegrads slektning har ESRD, er i følge et ennå upublisert materiale 7.2 (95 % CI 6.5-8.1). Alle diagnosekategorier er da inkludert i analysen. Når alle kjente hereditære nefropatier ekskluderes, er relativ risiko for å utvikle endestadie nyresvikt likevel betydelig forhøyet med relativ risiko på 3.7 (95% CI 3.1-4.4). Registerdata antyder at også andre typer nefropatier, som hypertensiv nefropati og interstitiell nefropati, akkumuleres i norske familier.
Det er i litteraturen beskrevet flere gener som øker risiko for ESRD (8–10). Felles for de fleste er at nedarvingen ikke følger tradisjonell autosomal dominant eller recessiv arvegang. Slike risikogener medfører som navnet sier, øket risiko for nyresvikt i en gitt situasjon. Men ofte må en annen risikofaktor for nyresvikt i tillegg være til stede, som f.eks diabetes mellitus. I enkelte tilfeller er det også beskrevet «gene on gene effect» (11), det vil si at et risikogen moduleres av et annet gen i nærheten. Det modulerende genet kan da gjøre risikogenet mer eller mindre risikabelt å inneha.
Etterhvert som vi får større kunnskap om genomet vårt og individuelle risikogen, øker også muligheten for ny, målrettet terapi. Selv om det pr. dags dato ikke eksisterer målrettet behandling for arvelig nyresykdom, er det vel verd å forsøke å finne ut om det foreligger nyresykdom hos flere personer i familien. Å oppdage fallende eGFR på et tidlig stadium, eller fange opp unge personer med familiær proteinuri, gjør at man i større grad kan komme tidlig til med behandling som modulerer allerede velkjente risikofaktorer for progress av nyresykdom. Man kan eventuelt komme tidlig i gang med antiproteinurisk behandling som ACE- hemmer eller angiotensin -2-blokker, hypertensjon kan behandles tidligere, og personer som ennå ikke har manifest nyresykdom, kan få råd om å unngå nefrotoksiske medikament.
Det finnes ingen kurativ behandling for ESRD. Selv nyretransplanterte pasienter har vanligvis redusert nyrefunksjon og følges nøye opp ved nefrologiske poliklinikker. Primærprofylakse og forebyggende tiltak kan forhåpentligvis bidra til at native nyrer lever lenger, og at behovet for nyreerstattende behandling utsettes.
Uromodulin (Tamm-Horsfall protein) er et velkjent protein som normalt finnes i urinen i små mengder, og som utøver en nyrebeskyttende effekt mot bl.a. infeksjoner. Ved mutasjoner i uromodulingenet (UMOD) endres proteinet og vil kunne gi skader i nyreparenkymet med interstitiell fibrose og progredierende nyresvikt. UMOD-assosiert nyresykdom er ikke særlig godt kjent, og er trolig en underdiagnostisert tilstand også i Norge. Tilstanden er også et godt eksempel på hvordan vi i dag bør involvere genetisk ekspertise på et tidligere stadium når vi mistenker arvelige nyresykdommer. Denne tilstanden følger et autosomalt dominant arvemønster, og vi har tidligere beskrevet diagnostikk og utredning av typiske forløp av sykdommen hos en mor (nyretransplantert) og hennes datter med hypertensjon, lett redusert nyrefunksjon, minimal proteinuri og ingen hematuri. Flere andre familiemedlemmer hadde også nyresykdom uten erkjent spesifikk diagnose (6). Det som tidligere var antatt å være to forskjellige sykdommer, har vist seg å være to ulike, men typiske, fenotyper av UMOD-mutasjoner. Den ene er medullær cystenyresykdom type II. Den andre er familiær hyperurikemi, ofte med typiske urinsyregiktanfall, som kan debutere allerede i tenårene. Hyperurikemi er et typisk tegn ved uromodulin-assosiert sykdom, og hos pasienter med redusert nyrefunksjon fra 30-40 års alder bør denne tilstanden vurderes. Ved hjelp av hel-genom analyser og genetiske koblingsanalyser kan man i dag ofte stille en eksakt diagnose. Den videre behandling kan konsentreres om å forebygge og/eller forsinke sykdomsprogresjon og komplikasjoner. God blodtrykkskontroll, profylakse mot urinsyregikt og tidlig behandling av forstyrrelser i kalsium-fosfat stoffskiftet vil være av stor betydning. Dette er også et eksempel på at betydningen av for eksempel nyrebiopsi i noen tilfeller vil kunne endres fra rent diagnostisk verdi til mer å karakterisere omfang av nyreskade og indikasjon for sykdomsdempende behandling.
Fabry sykdom er en X-bundet recessiv avleiringssykdom og har fått stor oppmerksomhet de senere år, særlig etter at farmasøytisk industri for vel 10 år siden syntetiserte det manglende enzymet, alfa-galaktosidase. Medikamentet gis som intravenøse infusjoner annenhver uke, oftest som hjemmebehandling. Slik enzymerstatningsterapi er svært kostbar, konferer oversikt over medikamentkostnader i Dagens Medisin sist høst (12). Prevalensen av sykdommen i Norge er større enn i mange andre land, og det er nylig erkjent at dette er en sykdom med mange flere fenotyper enn tidligere kjent. Sykdommen har således trolig en større prevalens enn tidligere antatt. Det kliniske spennet rangerer fra «klassisk» type hos menn (som ubehandlet medfører ca 20 års reduserte leveutsikter) til ikke-sykdomsgivende «non-klassiske» varianter av mutasjoner i alfa-galatosidase genet (13). De klassiske symptomene ved sykdommen er akroparestesier (i barneår), mens det i ungdomsår og voksen alder ofte tilkommer proteinuri og nyresvikt, kardiomyopati og/eller hjerneslag (gjerne i 30-40 års alder). Sykdommen er i Norge beskrevet med særlig stor utbredelse på Vestlandet (7), men det oppdages stadig flere tilfeller over hele landet. Særlig utfordrende er den nye erkjennelsen av «non-klassiske» mutasjoner med singel organ affeksjon eller fravær av klinisk sykdom. Konsekvensen av dette er sannsynligvis at mange av disse (de fleste?) ikke skal ha kostbar enzymbehandling. På den andre siden har vi nylig vist at pasienter med tidlig nyreaffeksjon kan oppnå regress av nyreskade med tidlig enzymbehandling (14). Screeningstudier på nyfødte i
enkelte land har vist en langt høyere prevalens av Fabry-mutasjoner enn tidligere erkjent (1:3000-4000). Betydningen av dette for senere sykdom er ukjent. Her gjenstår mye forskning. Det er viktig at praktiserende indremedisinere kjenner til diagnosen. Man skal særlig ha den i tankene når man står overfor pasienter med proteinuri/nyresvikt eller kardiomyopati uten kjent årsak, i særdeleshet dersom disse pasientene ikke har andre kjente risikofaktorer for kardiovaskulær sykdom, for eksempel vil Fabry-pasienter typisk ha et lavt blodtrykk. Ved mistanke om sykdommen henvises pasienten til nyreseksjonene ved Oslo universitetssykehus eller Haukeland universitetssykehus hvor kompetansesentrene for Fabry sykdom ligger.



