
Kristina H Haugaa
Av Kristina H Haugaa, Overlege, Ph.D, førsteamanuensis, Kardiologisk avdeling, Oslo Universitetssykehus, Rikshospitalet.
Plutselige, uventede dødsfall hos unge er alltid dypt tragisk og dramatisk. Hjertestans er den vanligste årsaken til plutselig, ikke-traumatisk død hos unge. Hos personer over 35 år er koronarsykdom den hyppigste årsaken til hjertestans, mens hos personer under 35 år skyldes det oftest arvelige hjertesykdommer.
Altfor ofte starter utredningen av familiær hjertesykdom etter at en tragedie har rammet et ungt individ. Utfordringen i kardiologien er å identifisere disponerte unge før plutselig død inntreffer. Fokus på familiære tilstander, familieanamnese og genetisk testing kan bidra til dette. Påvisning av en genfeil hos en pasient innebærer at slektningene kan undersøkes for familiens mutasjon, og man kan oppdage asymptomatiske familiemedlemmer som er genetisk disponert for sykdommen. Disse kan deretter tilbys profylaktisk behandling.
De arvelige hjertesykdommene deles inn i kardiomyopatier, med typiske strukturelle forandringer i hjertet som kan påvises ved billeddiagnostikk, og de elektriske arytmisyndromene eller ionekanalsykdommene som innebærer et strukturelt normalt hjerte. Felles for disse sykdommene er at de er arvelige og pasienter med disse sykdommene har økt risiko for hjerterytmeforstyrrelser og plutselig død. Alle tilstander som gjennomgås her er monogentisk arvelige og skyldes mutasjoner i gener som koder for proteiner i kardiomyocytten (Figur 1). Nedenfor følger en gjennomgang av de viktigste tilstandene.
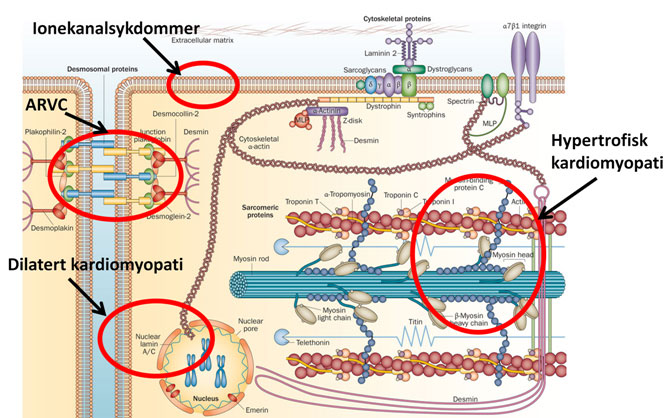
Figur 1. Figuren viser et utsnitt av en kardiomyocytt. De angitte cellestrukturenes spiller en rolle i patomekanismen for de ulike kardiomyopatiene. Modifisert etter Wilde AAM et al. 2013; Nat Rev Cardiol. Trykkes etter tillatelse.
ARVC er den hyppigste årsaken til plutselig død hos idrettsutøvere i Europa1. ARVC er autosomal dominant arvelig og skyldes mutasjoner i gener som koder for desmosomer, limet mellom cellene (Figur 1). Toppidrett fører til repetitivt stort mekanisk stress på hjertet, og svekkede desmosomer kan da føre til cellenekrose. De nekrotiske hjertemuskelcellene erstattes med fett og bindevev, og dette øker risikoen for livstruende ventrikulære arytmier. Høyt fysisk aktivitetsnivå kan utløse arytmier. Dessverre kan det første symptomet på ARVC være ventrikkelflimmer under fysisk aktivitet.
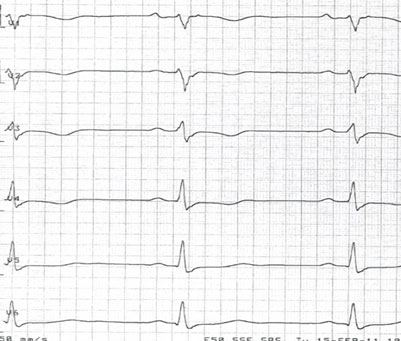
Figur 2 EKG fra en ARVC-pasient. Prekordiale avledninger V1-V4 viser typiske T-inversjoner.
Diagnosen ARVC stilles basert på funn fra EKG, Holter-monitorering, ekkokardiografi og MR-undersøkelse av hjertet2. På EKG ser man gjerne T-inversjon i prekordialavledningene (Figur 2), mens Holtermonitorering kan vise økt antall ventrikulære ekstraslag (>500 døgn).
På ekkokardiografi og MR ser man oftest en dilatert og tynnvegget høyre ventrikkel, men også venstre ventrikkel kan være affisert.
Gentest utføres hos pasienter med mistanke om ARVC, men et positivt svar må tolkes med forsiktighet på grunn av høy forekomst av genvarianter i normalbefolkningen3. Det er heller ikke slik at en negativ gentest utelukker ARVC.
Behandlingen baserer seg på antiarytmika (betablokker og amiodarone) i tillegg til en implanterbar hjertestarter (ICD) hos høyrisikopasienter. Pasienter med ARVC skal ikke drive konkurranseidrett.
HCM er den hyppigste årsaken til plutselig død hos idrettsutøvere i USA4. Sykdommen skyldes mutasjoner i gener som koder for sarkomerproteiner, det kontraktile apparatet i hjertemuskelcellene (Figur 1). Prevalensen er ca 1:500 og til tross for autosomal dominant arvegang er sykdommen hyppigere uttrykt hos menn enn hos kvinner. Venstre ventrikkelhypertrofi er definert som interventrikulærseptum > 1,5 cm uten annen forklarende årsak som for eksempel hypertensjon eller klaffefeil. HCM har variabel penetrans, det vil si familiemedlemmer med samme mutasjon kan ha alle grader av uttrykt sykdom, fra betydelig hypertrofi til helt normale funn.
Symptomer på HCM kan være dyspne, hjertebank eller rytmeforstyrrelser. Diagnosen stilles oftest ved hjelp av ekkokardiografi.
Gentest bekrefter diagnosen hos ca 50-60% av pasientene med HCM. Ved en positiv gentest kan man gå videre med genetisk testing av slektninger. Mutasjonspositive familiemedlemmer bør få utført en ekkokardiografi og følges videre hos kardiolog med jevne mellomrom. Hyppigheten av kontrollene er avhengig av alder, og hensikten er å fange opp hvis/når sykdommen progredierer.
HCM behandles med betablokkere og behovet for ICD vurderes på individnivå hos høyrisikopasienter. Risikofaktorer for plutselig død er synkoper, malign familiehistorie med plutselig død, septumtykkelse > 3cm, utilstrekkelig blodtrykksrespons ved arbeidsbelastning og arytmier på Holtermonitorering.
Hvis hypertrofien fører til utløpsobstruksjon, vurderes gradientreduserende behandling i form av myektomi (operativ fjerning av septumvalk) eller perkutan transluminal septum myokard ablasjon (PTSMA). PTSMA innebærer et kateterindusert infarkt i septum som fører til krymping av septumvalken.
Pasienter med HCM anbefales ikke å drive konkurranseidrett5.
Dilatert kardiomyopati er arvelig i 30-50 % av tilfellene og kan debutere i ung alder. Vanligvis kommer pasientene initialt med hjertesviktsymptomer, men arytmier kan også være det første symptomet. Diagnosen stilles oftest på bakgrunn av ekkokardiografi. Ved familiær dilatert kardiomyopati kan gentest vurderes.
Spesiell oppmerksomhet må rettes mot pasienter med en kombinasjon AV-blokk, atrieflimmer og ventrikulære takykardier. Disse kan ha underliggende mutasjoner i Lamin A/C-genet og dette gir ofte en malign form for dilatert kardiomyopati med høy hyppighet av plutselig død6. Laminproteiner er viktige byggesteiner for cytoskjelettet og cellestrukturen (Figur 1). Ofte debuterer disse pasientene med arytmier som for eksempel atrieflimmer eller AV-blokk rundt 40-års alder. Behandlingen er som for dilatert kardiomyopati av annen årsak inkludert hjertesviktmedikasjon og antiarytmisk behandling. Hos pasienter med Lamin A/C mutasjoner bør ICD vurderes på et tidligere tidspunkt enn ved annen genese på grunn av den høye forekomsten av maligne arytmier.
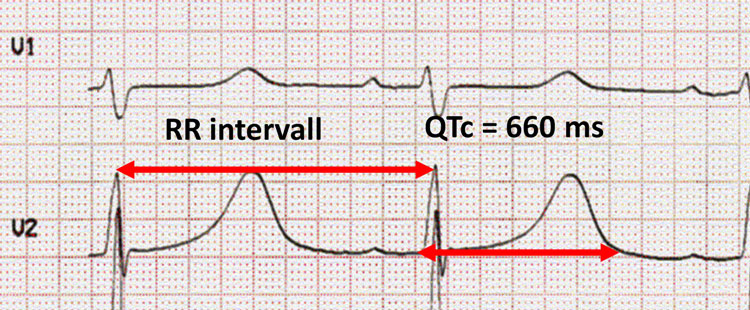
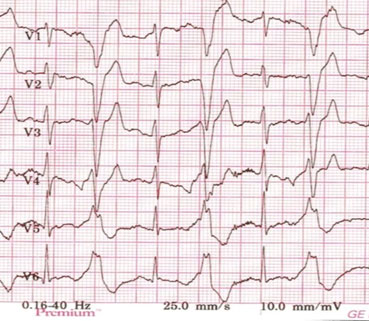
Figur 3. EKG fra en pasient med forlenget QT-tid.
LQTS ble første gang beskrevet av de to norske legene Anton Jervell og Fred Lange-Nielsen, som i 1957 skrev om en familie der 4 av 6 barn var døve og hadde lang QT-tid på EKG7. Tre av de døve barna døde tidlig, før 8-års alder. Romano og Ward beskrev senere et liknende symptomkompleks, men uten døvhet8. Syndromene ble oppkalt etter forfatterne. I 1991 fant man den genetiske linken mellom disse syndromene; det viste seg at de med Jervell og Lange-Nielsen syndrom var de homozygote mutasjonsbærere og de med Romano-Ward syndrom var heterozygote mutasjonsbærere for mutasjoner i gener som koder for kardiale ionekanaler. Det er i dag kjent en rekke ulike gener der mutasjoner kan føre til LQTS, men typene LQT1, LQT2 og LQT3 er fortsatt de hyppigste. Ionekanaldefektene fører til et forlenget kardialt aksjonspotensial og dette kan avleses i EKG som forlenget QT-tid (Figur 3). Forlenget QT-tid disponerer for polymorfe ventrikulære arytmier, typisk Torsade de pointes-arytmien som oversatt betyr «danse rundt en linje» (Figur 4).
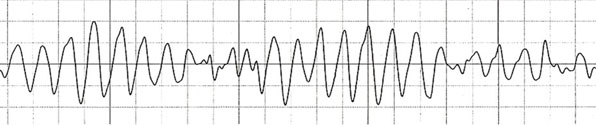
Figur 4, Torsade de pointes ventrikulær arytmi.
Synkoper er typiske symptomer, og disse er ofte trigget av fysisk anstrengelse eller psykisk stress og debuterer hyppigst i barneårene. Svømming har vist seg å være en klassisk trigger for arytmier og LQTS må vurderes ved drukning/ nærdrukningsulykker hos svømmedyktige.
Diagnosen stilles ved typiske symptomer med adrenergt utløste synkoper og forlenget QT-tid på EKG. QT-tiden måles fra starten av Q-bølgen til slutten av T-bølgen. QT-tiden varierer med hjertefrekvensen og skal derfor frekvenskorrigeres (QTc). Mest brukt er Bazetts formel som gir QTc = QT/ √RR intervallet (målt i sekunder). Tradisjonelle normalverdier for QTc har vært < 450 ms hos menn og < 460ms hos kvinner, men dette fører nok til en del overdiagnostisering, og det er foreslått nye grenser der QTc < 480 ms er innenfor normalvariasjonen. Det er viktig å merke seg at QTc > 500ms er klart forlenget og utgjør en risiko for maligne ventrikulære arytmier.
Gentesting er nyttig hos pasienter med forlenget QT-tid. Det kan bekrefte diagnosen og gir tilleggsinformasjon i form av genotype (LQT1, LQT2 osv) som er viktig for risikovurdering og optimal behandling.
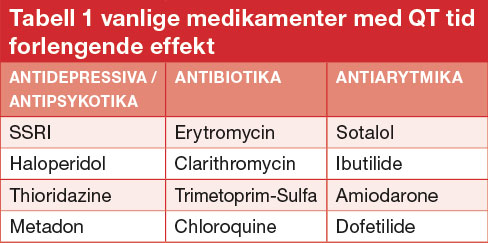
Mange vanlige legemidler forlenger QT-tiden og dette er den hyppigste medisinske årsaken til at et medikament blir trukket fra markedet. Aktuelle medikamenter som forlenger QT-tiden er antibiotika, anti-depressiva og antipsykotika. En oppdatert liste over disse medikamentene finnes på www.CredibleMeds.org.
Pasienter med LQTS må instrueres i å unngå QT-tid forlengende medikamenter da dette øker deres risiko for arytmier. Videre skal de ikke drive konkurranseidrett. Medikamentell behandling er basert på høydose betablokkermedikasjon. I noen tilfeller er det nødvendig med ICD-implantasjon.
Et vanlig scenario ved indremedisinske avdelinger er eldre, multimorbide pasienter med polyfarmasi. Dette disponerer for ervervet lang QT-tid og maligne arytmier. Dette er vanligvis ikke en arvelig tilstand, men prinsippene er de samme som for LQTS. Mange medikamenter blokkerer de ionekanaler som er ansvarlige for den kardiale repolariseringen og derved imiteres den medfødte sykdommen LQTS. Risiko for ervervet lang QT-tid og Torsade de pointes arytmi er økt hos følgende pasientgrupper, og risikoen øker med antall risikofaktorer9.
– Bruk av QT-tid forlengende medisiner
– Kvinne
– Hypokalemi
– Bradykardi
– Komorbiditet (hjertesvikt, nyresvikt, leversvikt)
Mange vanlige medikamenter har QT tid forlengende effekt slik som antibiotika (makrolidantibiotika), antidepressiva og antiarytmika (Tabell 1). Ved ervervet lang QT-tid > 500 ms må utløsende faktorer gjennomgås. Man bør seponere QT-tid forlengende medikamenter, de aller fleste lar seg erstatte med et medikament uten slik risiko. Videre må elektrolyttforstyrrelser korrigeres, og pasienten må instrueres i hva de skal gjøre i risikosituasjoner for eksempel ved langvarige diaréer med ledsagende dehydrering og hypokalemi. Hos pasienter med medikamentindusert torsade de pointes arytmi, kan gentesting med tanke på underliggende LQTS vurderes, da det er angitt at ca 15% kan ha underliggende mutasjon i et LQTS-relatert gen.
Figur 5, Bigemini ved arbeids-EKG. Typisk eksempel på ventrikulære ekstrasystoler i bigemini som opptrer ved fysisk belastning hos en pasient med CPVT.
Katekolaminerg polymorf ventrikkeltakykardi (CPVT) er en sjelden, autosomal dominant arvelig hjertesykdom som predisponerer for ventrikkeltakykardi ved adrenerge stimuli. Ved CPVT foreligger en defekt Ryanodinreseptor. Ryanodinreseptoren er vikitg for den intracellulære kalsiumreguleringen. Pasienter med CPVT får typisk økende ventrikulære ekstraslag under belastning som så kan slå over i VT og i verste fall VF og gi plutselig død. Klassiske funn ved anamnese er synkoper som inntreffer ved fysisk aktivitet eller emosjonelt stress og som gjerne debuterer i barneårene. Igjen er også svømming en viktig trigger for arytmi.
Diagnosen stilles ved arbeids-EKG der man påviser økende ventrikulær ekstrasystoli10 (Figur 5) Hvile-EKG er oftest helt normalt. Gentest bør utføres ved sterk mistanke om CPVT og pasienten kan henvises til spesialisert senter ved klar mistanke om CPVT eller ved etablert diagnose.
Behandlingen består av høydose betablokkermedikasjon. ICD må vurderes individuelt.
Genetiske undersøkelser med tanke på kardiomyopatier og kardiale kanalopatier utføres i Norge ved Enhet for hjertegenetikk, Oslo Universitetssykehus, Ullevål.
Det man trenger for analyse er et glass med EDTA-blod. Pasienter med mistanke om genetisk hjertesykdom bør få utført gentest på klinisk indikasjon. Det bør alltid foreligge en klar fenotype før man starter gentesting. Det kan med fordel konfereres med et spesialisert senter for genetiske hjertesykdommer med tanke på indikasjonsstilling.
Det er enighet om at man for de ovenstående sykdommene har dekning for presymptomatisk genetisk testing og familiescreening. Dette innebærer at familiemedlemmer til en pasient med en genetisk bekreftet hjertesykdom, kan velge om de vil undersøkes for å se om de er bærere av familiens mutasjon. Før presymptomatisk testing utføres må de pårørende motta genetisk veiledning for å få informasjon om hva et eventuelt mutasjonsfunn vil innebære for dem både medisinsk, men også praktisk for eksempel med tanke på egne forsikringer. Genetisk veiledning utføres ved de respektive sykehusenes genetiske avdelinger. Familiemedlemmer som tester positivt for familiens mutasjon må følges opp kardiologisk med tanke på risikostratifisering, om de utvikler sykdommen og eventuell behandling.
Tilgjengeligheten av genetisk testing øker i stor fart. Det finnes en rekke internasjonale og også nasjonale tilbydere der man enkelt kan bestille en gentest på internett. Testen er da ofte basert på analyse av en spyttprøve eller vattpinneprøve fra munnslimhinnen. Her kan man rekvirere alt fra farskaps-, søskenskaps-, og tvillings-tester til risikoprofiler for diverse sykdommer og plutselig død. Man må ha i mente at disse testene har varierende grad av kvalitetssikring og tolkningen av testene er ikke alltid enkle. Helt sikkert er at vi kommer til å se mer, lære mer og bli nødt til å tolke mer genetisk informasjon i fremtiden.
Referanser



