
Kaspar Broch
Av Kaspar Broch, forsker og lege i spesialisering, kardiologisk avdeling, Oslo Universitetssykehus, Rikshospitalet
Ved dilatert kardiomyopati utvides og svekkes (i hovedsak venstre) hjertekammer, uten at koronarsykdom eller patologisk hemodynamisk belastning er årsaken. Tilstanden er det felles fenotypiske uttrykket for en rekke, ulike patogenetiske prosesser som affiserer selve myokard. Når svekkelsen av myokard overskrider hjertets kompensasjonsevne, utvikles symptomer på hjertesvikt. Behandlingen er som ved hjertesvikt av annen årsak. Prognosen er bedre enn ved iskemisk kardiomyopati.
Dilatert kardiomyopati er karakterisert ved utvidelse av og redusert systolisk funksjon av venstre ventrikkel i fravær av koronar, kongenitt, valvulær eller hypertensiv etiologi1, jf. figur 1. Denne europeiske definisjonen tar utgangspunkt i det fenotypiske uttrykket, som igjen har en rekke forskjellige årsaker. Den amerikanske klassifikasjonen tar utgangspunkt i etiologien og skiller primært mellom arvelige og ikke-arvelige kardiomyopatier.2
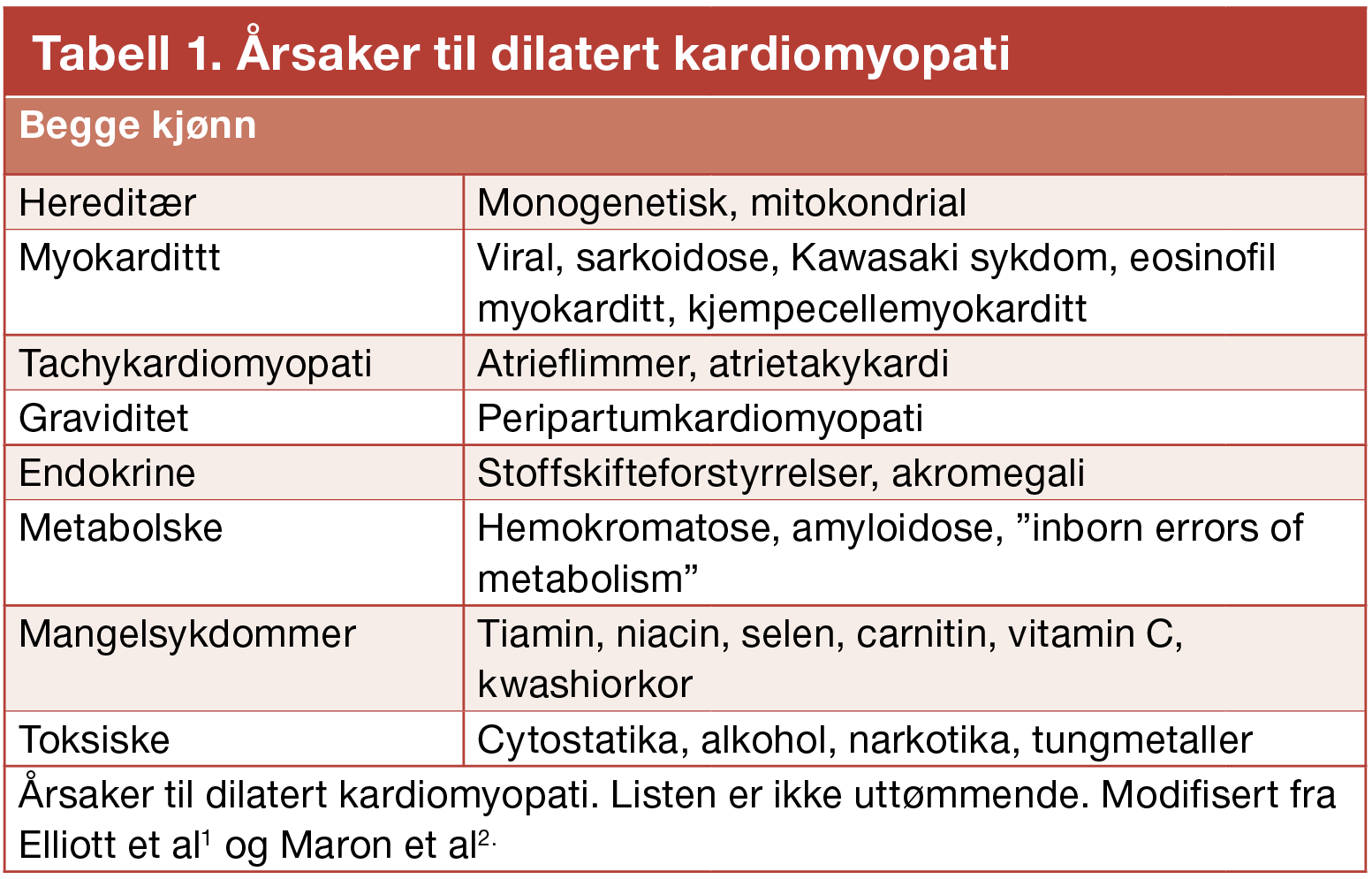
Årsaker til dilatert kardiomyopati er listet opp i tabell 1. I de fleste tilfellene klarer man ikke avdekke noen spesifikk etiologi, og tilstanden betegnes ”idiopatisk” dilatert kardiomyopati. Man tror at en stor andel (30 – 50 %) av disse tilfellene er hereditære, men på grunn av inkomplett penetrans og variabelt fenotypisk uttrykk kan arvelighet være vanskelig å fastslå.3 Mutasjon i de genene som så langt er fastslått å være involvert i patogenesen utgjør kun et fåtall av de antatt familiære tilfellene.4 En annen årsak til dilatert kardiomyopati antas å være gjennomgått eller persisterende myokarditt.5 Viktige, dels reversible, årsaker inkluderer langvarig takykardi, systemiske inflammasjonssykdommer, alkoholisme og hemokromatose. Uavhengig av utløsende årsak reagerer kroppen på den reduserte hjerte-pumpefunksjonen med en rekke kompensatoriske mekanismer. Disse inkluderer nevrohormonal aktivering med økt aktivitet i renin-angiotensin-aksen og det sympatiske nervesystem. Selve hjertet gjennomgår en såkalt remodelleringsprosess der venstre hovedkammer utvides, hjertemuskelceller dør og erstattes med bindevev, og den ekstracellulære matrixen endrer sin sammensetning. På det ultrastrukturelle plan tilkommer økt transkripsjon av føtale isoformer av kontraktile proteiner og enzymer som inngår i cellemetabolismen.
Dilatert kardiomyopati antas å være årsaken til ca 10 % av hjertesvikttilfellene i vår del av verden.6 Typisk er pasientene yngre og har mindre komorbiditet enn den gjennomsnittlige pasienten med iskemisk betinget hjertesvikt eller hjertesvikt med bevart ejeksjonsfraksjon. Ofte er symptomutviklingen snikende, og en stor andel av pasientene har vært feildiagnostisert med astma eller luftveisinfeksjon før de legges inn i sykehus med forverring av hjertesvikten. Hos noen pasienter ses et mer dramatisk forløp med rask utvikling av fulminant hjertesvikt. Symptomene er i første rekke funksjonsdyspnoe, sjeldnere hjertebank, synkope eller hjertestans på grunn av ventrikulær arytmi. Kliniske funn kan inkludere kompensatorisk tachycardi, systolisk bilyd (p.g.a. mitralinsufficiens sekundært til dilatasjon av venstre ventrikkel), krepitasjoner over lungene, hals-venestase og perifere ødemer. Tidlig i forløpet er det ofte ingen patolog-iske funn ved klinisk undersøkelse!
Diagnosen må først og fremst mistenkes hos pasienter med funksjonsdyspnoe eller redusert arbeidstoleranse uten annen forklaring. Familieanamnese er viktig. Differensialdiagnostisk kan oppgangen mot lungesykdommer være vanskelig. EKG er ofte uspesifikt, men sjelden normalt. Lungefunksjonsverdier som FEV1 og FVC vil ofte være lett nedsatte p.g.a. lungestuvning og stort hjerte, men sjelden i den grad at de forklarer pasientens symptomer. B-type natriuretiske peptider (BNP) vil kunne hjelpe i den diagnostiske utredningen: Normal BNP/NT-proBNP nærmest utelukker hjertesvikt. Rtg thorax vil i de fleste tilfeller vise forstørret hjerte og eventuelt lungestuvning. Endelig diagnose stilles ved ekkokardiografi i kombinasjon med koronarangiografi og krever at det er dilatasjon og svekkelse av venstre ventrikkel som ikke kan forklares av koronarsykdom, primær klaffesykdom eller antatt langvarig hypertensjon. Inflammatorisk systemsykdom som SLE og Wegners granulomatose og metabolsk sykdom som hemokromatose er sjeldne årsaker som kan behandles og derfor må utelukkes. Videre utredning bør skreddersys den enkelte pasient. Hos ca. 10 % vil man kunne avdekke en arvelig årsak ved genetisk screening. Disse pasientene må tilbys genetisk veiledning. MR-undersøkelse med kontrast kan avdekke inflammatorisk årsak. Ved rask hjertesviktutvikling som ikke lar seg reversere ved hjelp av medikamenter, eller som ledsages av ventrikulære arytmier, er det viktig å avdekke eventuell kjempecellemyokarditt eller eosinofil myokarditt, som krever spesifikk behandling og har svært dårlig prognose ubehandlet. Disse pasientene bør biopseres. Forøvrig bør hjertebiopsi kun utføres ved mistanke om spesifikk tilgrunnliggende årsak.
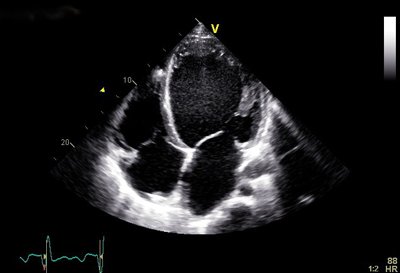
Figur 1. Dilatert kardiomyopati. Ekkokardiografisk apikalt firekammerbilde av en pasient med dilatert kardiomyopati. Venstre ventrikkel (VV) er utvidet og sfærisk.
Eventuelle reversible årsaker som takykardi, alkoholisme, inflammatorisk systemsykdom, sarkoidose og hemokromatose må avdekkes og behandles. Pasienter med hereditær årsak kan tilbys genetisk veiledning og eventuelt genetisk kaskadeundersøkelse av familiemedlemmer. For øvrig skiller ikke behandlingen ved dilatert kardiomyopati seg fra behandlingen ved hjertesvikt generelt, der diuretika, hemmere av renin-angiotensin-aldosteronaksen og betablokkade står sentralt. Ved ejeksjonsfraksjon under 35 % og QRS-bredde > 150 ms er det indikasjon for resynkronsieringsterapi, og pasienter med ejeksjonfraksjon < 35 % bør inividuelt vurderes med tanke på primærprofylaktisk hjertestarter. Effekten av retningslinjebasert hjertesviktbehandling er ofte formidabel hos pasienter med dilatert kardiomyopati og overskrider den man ser ved for eksempel hjertesvikt etter gjennomgått hjerteinfarkt. Hos et fåtall av pasientene progredierer sykdommen mot terminal hjertsvikt. Disse pasientene er ofte gode kandidater for hjertetransplantasjon.
Prognosen ved hjertesvikt er generelt dårlig, men avhenger av årsaken. Prognosen ved idiopatisk dilatert kardiomyopati er bedre enn ved koronariskemisk hjertesvikt .7 Mye tyder på at overlevelsesutsiktene har bedret seg over de siste tiårene med bedret hjertesviktbehandling.8,9 I en prospektiv kohortstudie som nylig ble utført på 102 pasienter med dilatert kardiomyopati som var henvist til Rikshospitalet for utredning av hjertesvikt, opplevde flertallet av pasientene en betydelig symptomatisk bedring i løpet av et år etter diagnostisk utredning og igangsatt, retningslinjebasert hjertesviktbehandling.10 Venstre ventrikkels ejeksjonsfraksjon steg fra 26 ± 10 % til 40 ± 11 % i løpet av dette året. Etter median 3,6 års oppfølging var ni pasienter transplantert og fire døde. Fem års transplantasjonsfri overlevelse var 84 %.11
Dilatert kardiomyopati er i dag en sekkediagnose for enhver tilstand som direkte affiserer myokard og induserer dilatasjon og nedsatt systolisk funksjon av venstre ventrikkel. Med bedret forståelse av underliggende genetikk og inflammatorisk patogenese vil man forhåpentlig i fremtiden kunne tilby bedret etiologisk diagnostikk. Som følge av dette vil diagnosen ”idiopatisk dilatert kardiomyopati” fragmenteres, og forhåpentlig vil man etter hvert kunne tilby pasientene skreddersydd behandling rettet mot den underliggende årsaken.
Referanser



