
John Willy Haukeland
John Willy Haukeland, PhD, spesialist i generell indremedisin og fordøyelsessykdommer, overlege, Gastromedisinsk avdeling, OUS Ullevål
Det kliniske bildet ved kroniske autoimmune leversykdommer er heterogent, og beror i stor grad på om sykdomsprosessen rammer leverparenchymet eller galleveiene, og om det utvikler seg levercirrhose, portal hypertensjon og malignitet. Autoimmune leversykdommer medfører ofte betydelig sykelighet, og er en viktig årsak til levertransplantasjon.
Autoimmun sykdom kan inntreffe når balansen mellom immuntoleranse og immunreaktivitet forrykkes mot immunreaktivitet. Leveren regnes som et relativt immuntolerant organ, noe som gjenspeiles i at behovet for immunsuppresjon etter levertransplantasjon ofte er mindre enn ved andre organtransplantasjoner. En viss grad av immuntoleranse er antagelig viktig for at leveren skal kunne rense portveneblodet uten at immunforsvaret blir «uhensikts-messig aktivert». Men eksponeringen for antigene stimuli er kontinuerlig til stede, og autoimmun sykdom kan derfor også ramme leveren (1).
Autoimmun hepatitt (AIH), primær biliær cirrhose (PBC) og primær scleroserende cholangitt (PSC) er kroniske leversykdommer som oppfattes som autoimmune. Argumenter for dette er gjengitt i tabell 1.
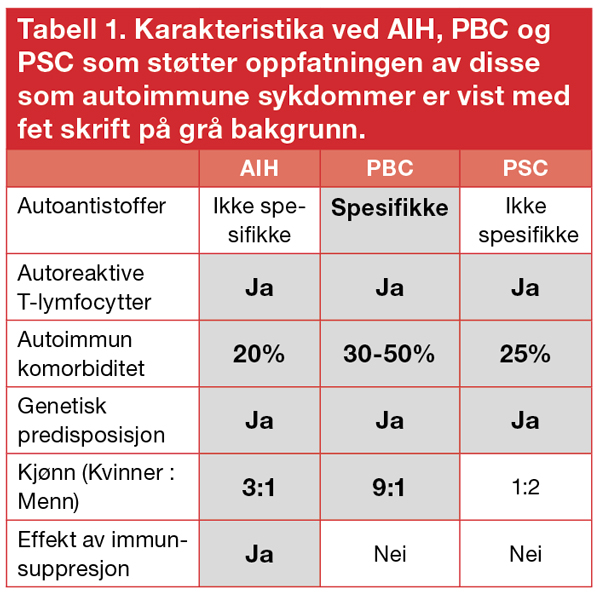
Årsaken til disse sykdommene er ikke kjent, men genetisk disposisjon i kombinasjon med utløsende miljøfaktorer spiller antagelig en avgjørende rolle. Forekomsten er relativt lav (Tabell 2) (2).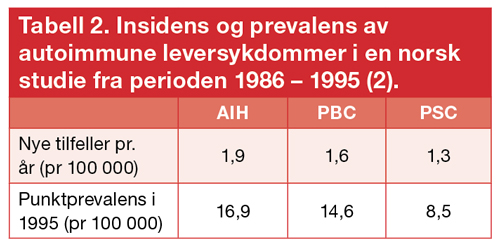
Nedenfor presenteres disse tre sykdommene separat, men i praksis er det kliniske bildet ikke alltid like tydelig. Noen ganger ses overlapp mellom AIH og PBC eller AIH og PSC. I denne artikkelen omtales ikke IgG4-relatert sykdom, en «ny» steroidresponsiv sykdomsentitet som bl.a. kan ramme pancreas, galleveier og lever. Hepatisk sarkoidose og hepatisk affeksjon ved andre revmatologiske sykdommer omtales heller ikke.
Ved mistanke om kronisk leversykdom er autoimmun leversykdom alltid en av flere aktuelle differensialdiagnoser. Hvis det samtidig foreligger andre funn som passer med disse sykdommene, vil mistanken skjerpes; f.eks er AIH assosiert med leddsmerter og generell sykdomsfølelse, PBC med asteni og kløe mens PSC er assosiert med inflammatorisk tarmsykdom. Andre leversykdommer er dog vanligere, og utredningen må som et minimum alltid avklare om det foreligger alkoholisk leversykdom, nonalkoholisk fettlever (NAFLD), kronisk viral hepatitt eller hemokromatose.
Kroniske leversykdommer er ofte asymptomatiske inntil det foreligger cirrhose. Derfor bør også pasienter uten symptomer utredes når kliniske funn, blodprøver eller billeddiagnostikk gir mistanke om leversykdom.
Man må skille mellom «skade-markører» (ASAT, ALAT, ALP, GT) og «funksjonsmarkører» (INR, albumin, bilirubin). Funksjonsmarkørene blir som regel ikke patologiske før det foreligger avansert sykdom. Skademarkørene derimot, vil som regel være patologiske allerede tidlig i sykdomsprosessen, men ikke nødvendigvis veldig uttalt, og i perioder vil de også kunne være innenfor normalområdet. Ved AIH stiger primært ASAT og ALAT som uttrykk for hepatocyttskade. Ved PBC og PSC stiger primært ALP og GT som uttrykk for skade i galleveiene. Ved langtkommen sykdom vil skademarkørene kunne normaliseres som følge av tap av gjenværende leverparenchym eller fordi sykdomsprosessen brenner ut.
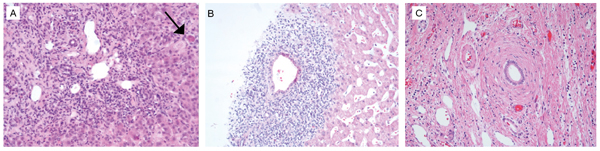
Figur 1. Leverbiopsier ved autoimmune leversykdommer. A) Autoimmun hepatitt; betennelsesinfiltrat i portalområdet som strekker seg innover i leverlobuli (interfaseaktivitet) og nekrotisk hepatocytt (pil), B) Primær biliær cirrhose; betennelsesinfiltrat i og omkring en gallegang, C) Primær scleroserende cholangitt; konsentrisk fibrose omkring en gallegang. Bildene er stilt til rådighet av Dr. Løberg (A,B) og Dr. Reims (C), Oslo universitetssykehus.
Klinisk presentasjon
Det kliniske bildet ved AIH er mangfoldig. Hos et mindretall manifesterer sykdommen seg som en akutt hepatitt med ikterus, kvalme, anorexi og nedsatt almentilstand. De fleste blir imidlertid diagnostisert i asymptomatisk fase eller med uspesifikke symptomer som f.eks artralgier, noe som er relativt vanlig. Ettersom sykdommen ofte utvikler seg uten spesielle symptomer, har hele 30% rukket å utvikle cirrhose på diagnosetidspunktet.
Hvordan settes diagnosen?
AIH skyldes en cellemediert immunrespons rettet mot levervevet. Leverbiopsi er nødvendig for diagnosen. Typisk ses inflammatorisk aktivitet i portalområdene som sprer seg innover i leverlobuli (Figur 1A), ofte ses økt mengde plasmaceller. Det histologiske bildet vil kunne støtte diagnosen, men er ikke patognomonisk.
Diagnosen baserer seg også på funn av moderat eller høyt titer av visse autoantistoffer slik som glatt muskel antistoffer (SMA), kjerneantistioffer (ANA) og/eller høye IgG verdier. Andre autoantistoffer som er assosiert med AIH er anti-LKM-1 (liver kidney-microsome) og anti-SLA (soluble liver antigen). Positiv SMA og ANA i lavt titer er vanlig ved kroniske leversykdommer, ikke minst ved NAFLD (3), og skal ikke tillegges vesentlig vekt. I tilfeller der diagnosen er usikker, vil en klar biokjemisk respons etter et behandlingsforsøk med steroider kunne styrke diagnosen. Ved samtidig kronisk viral hepatitt vil det i praksis være vanskelig å si om det også foreligger AIH. I slike tilfeller må det først gis antiviral behandling, før man eventuelt kan konkludere med AIH.
Behandling
Ubehandlet er AIH en potensielt alvorlig sykdom med økt risiko for levercirrhose og død. Immundempende behandling bedrer prognosen, de fleste vil derfor ha normale leveutsikter når behandling innsettes tidlig. Men ved behandlingssvikt, og når diagnosen stilles sent i forløpet, vil det kunne inntreffe betydelig sykelighet og livstruende komplikasjoner.
Behandling ved AIH er godt beskrevet i europeiske retningslinjer (4). Målet med behandlingen er å indusere komplett remisjon og dermed forhindre videre sykdomsprogresjon. Indikasjon for behandling er klart til stede når det foreligger histologisk bekreftet inflammasjon utover det helt minimale og alltid i tilfeller med avansert fibrose. Selv ved etablert cirrhose er det riktig å tilby behandling da tilstanden kan stabiliseres, men ved dekompensert cirrhose må man avstå fra immunsuppresjon da risikoen for komplikasjoner blir større enn den potensielle gevinsten.
I tilfeller der transaminasene kun er lett forhøyet (< 2 x øvre normalverdi) samtidig som det bare foreligger minimal histologisk sykdomsaktivitet, kan en avventende holdning forsvares. Man må da tilby langtidsoppfølging med blodprøver 2-4 ganger pr år for å fange opp en eventuell økt sykdomsaktivitet som krever behandling.
Standard induksjonsbehandling er prednisolon 0,5-1 mg/kg. De aller fleste oppnår med dette klinisk og biokjemisk respons med tydelig fall av transaminaser i løpet av dager/uker. Fall i IgG, som korrelerer godt med sykdomsaktivitet, kommer noe senere. Det anbefales å legge til azatioprin (1-2 mg/kg) etter noen uker samtidig som prednisolondosen reduseres. Dosering av begge medikamenetene individualiseres og styres etter responsen. Noen trenger langtidsbehandling med både prednisolon og azatioprin, men mange kan klare seg med kun et av disse medikamentene. Hvilket medikament som velges må avgjøres sammen med pasienten, og man må ta hensyn til bl.a. alder, komorbide tilstander, risiko for medikament-bivirkninger m.m. Budesonid, et kortikosteroid med mindre systemiske effekter, er et godt alternativ til prednisolon hos dem som ikke har utviklet cirrhose. Hvis man ikke oppnår full remisjon med ovenstående medikamenter, er annen immunsupresjon aktuelt, f.eks mycofenolat mofetil eller tacrolimus.
Seponering av behandling
AIH er en kronisk sykdom der 80-90% får tilbakefall ved seponering av immunsupresjon, som regel innen et år. Det kan likevel være viktig å gjøre seponeringsforsøk hos enkelte, spesielt der det ikke foreligger avansert leversykdom. Seponeringsforsøk anbefales tidligst etter tre år, og kun hvis ny leverbiopsi bekrefter histologisk remisjon og hvis det har vært biokjemisk normalisering i minst to år. Ved eventuell seponering må pasienten følges tett for å fange opp tilbakefall.
Klinisk presentasjon
PBC skyldes en inflammatorisk prosess rettet mot de små, intrahepatiske galleganger. Sykdommen rammer i all hovedsak kvinner. PBC er sjelden før 30 års alder, men kan ramme alle aldersgrupper etter det. Kardinalsymptomene er asteni og kløe, men symptomene er ofte vage, og mange diagnostiseres uten å ha disse plagene eller andre symptomer på leversykdom. Diagnosen settes ofte før det har utviklet seg cirrhose. Betegnelsen primær biliær cirrhose er derfor inadekvat og i dag anvendes betegnelsen primær biliær cholangitt i økende grad. Mellom 30 og 50% av pasientene har andre autoimmune sykdommer, de vanligste er Sjøgrens syndrom og Hashimotos thyroiditt (5).
PBC er en cholestatisk sykdom der kolesterol- og gallesyremetabolismen er påvirket. Det ses ofte hyperkolesterolemi, men uten at den kardiovaskulære risiko nødvendigvis er økt. Ved avansert sykdom kan man se mangel på fettløselige vitaminer, deriblant vitamin D, noe som er med på å forklare økt forekomst av osteoporose ved PBC, men her spiller også andre faktorer en rolle.
Hvordan stilles diagnosen?
Autoantistoffer rettet mot mitokondrier (AMA) er en svært sensitiv markør for PBC idet om lag 95% er positive. Samtidig er AMA relativt spesifikk. Ved forhøyet ALP uten annen forklaring (f.eks. beinsykdom eller ekstrahepatisk obstruksjon av galleveiene) er påvisning av AMA i titer ≥1/40 diagnostisk for PBC. Mange har også høy IgM, og noen har påvisbare kjerneantistoffer (ANA).
Positiv AMA kan også ses hos pasienter uten PBC, men kan da betraktes som en risikomarkør for fremtidig PBC (20% innen 5 år) (6).
Biopsi anses ikke nødvendig for diagnosen, men er nyttig for å kartlegge grad av fibrose, og for å se om det foreligger overlapp mot AIH eller andre leversykdommer som f.eks. steatohepatitt. Ved PBC ses inflammatorisk infiltrat rundt galleveiene (Figur 1B), av og til med granulomer.
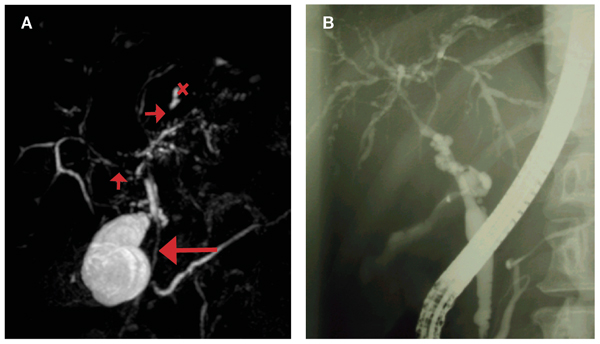
Figur 2. Cholangiogram ved primær scleroserende cholangitt fremstilt ved MRC (A) og ved ERC (B) (to forskjellige pasienter). På MRC-bildet ses bortfall av galleganger (korte piler), stent i en stenotisk choledochus (lang pil) og prestenotisk dilatasjon (X). På ERC-bildet ses klassisk «kaliberveksling» av intrahepatiske galleganger. Bildene er stilt til rådighet av Dr. Hanekamp (A) og Dr. Aabakken (B), Oslo universitetssykehus.
Behandling
Langtidsbehandling med gallesyren ursodeoksykolsyre, 13-15 mg/kg/dg, forsinker sykdomsprogresjonen og øker transplantasjonsfri over-levelse (7). Med mindre det foreligger avansert sykdom, vil pasienter som responderer på behandlingen ha leveutsikter lik alders- og kjønnsmatchete kontroller. Ursodeoksykolsyre er relativt vannløselig, noe som medfører at «gallen flyter lettere». I tillegg har denne gallesyren antiinflammatoriske egenskaper. Nord-amerikanske svartbjørn har høye nivåer av ursodeoksykolsyre, spesielt i vinterhalvåret, og dette er sannsynligvis en viktig fysiologisk adapatasjon som muliggjør bjørnens lange vinterdvale (8).
40 % er imidlertid non-respondere på ursodeoksykolsyre, og for disse har vi per i dag ingen effektiv behandling. Immunsupresjon har ingen effekt ved PBC. I en fersk studie med obetikolsyre så man biokjemisk respons hos om lag halvparten av non-respondere (9), men det er foreløpig uavklart hvilken plass obetikolsyre skal ha i behandlingen ved PBC ettersom langtidsstudier mangler. Virkningsmekanismen ved obetikolsyre beror på dens agonistiske effekt på farnesoid-X reseptor noe som resulterer i økt gallesekresjon og nedregulering av proinflammatoriske og profibrotiske signalveier.
Kløe er vanlig ved PBC og kan noen ganger utgjøre et betydelig problem. Ursodeoksykolsyre hjelper ikke. Gallesyrebinderen cholestyramin og antibiotikumet rifampicin kan ha gunstig effekt, men disse behand-lingene har flere problematiske bivirkninger. Antihistaminer brukes mye, men effekten er dårlig dokumentert. Lysbehandling kan være nyttig.
Ulike grader av asteni er vanlig ved PBC og er i liten grad relatert til sykdomsstadiet. Hos noen medfører dette en betydelig funksjonsned-settelse. Hverken ursodeoksykolsyre eller annen medikamentell terapi har vist effekt i forhold til dette.
Leverenheten ved OUS Rikshospitalet og senere Norsk senter for PSC har i en årrekke gjennomført omfattende forskningsaktivitet som har bidratt til økt kunnskap om PSC (10, 11).
Klinisk presentasjon
Pasienter med PSC er ofte unge (mellom 20 og 40 år). Sykdommen rammer menn oftere enn kvinner (2:1). 85 % av pasientene har inflammatorisk tarmsykdom (IBD). Motsatt, blant pasienter med IBD, ses PSC hos 5-10%.
Ved PSC pågår en inflammatorisk/fibrøs prosess omkring ekstra- og intrahepatiske galleveier. Dette resulterer i strikturer i galletreet som disponerer for episoder med icterus og ascenderende cholangitter. Mange plages med kløe. Alvorlighetsgraden er ulik fra pasient til pasient, og sykdomsbildet karakteriseres av stor variasjon og uforutsigbarhet.
Den inflammatoriske prosessen forårsaker etter hvert økende leverfibrose. Noen utvikler lever-cirrhose. Det at fibrosedannelsen har sitt utspring i portalområdene, og ikke i parenchymet, medfører at portal hypertensjon kan inntreffe før det foreligger biokjemiske tegn til nedsatt leverfunksjon og i noen tilfeller før det foreligger levercirrhose.
Cholangiocarcinom er en fryktet komplikasjon av PSC. Livstidsrisikoen for dette er så høy som 10-20%. Mange er dessverre inoperable når diagnosen stilles, og prognosen er da dårlig med meget lav 5-årsoverlevelse.
Hvordan stilles diagnosen?
Diagnosen baseres på kombinasjonen kronisk forhøyede staseparametre (ALP, GT) og kalibeveksling av galletreet enten fremstilt ved magnetisk resonans cholangiografi (MRC) (Figur 2A) eller ved endoskopisk retrograd cholangiografi (ERC) (Figur 2B). Positivt funn ved MRC er vanligvis tilstrekkelig for diagnosen, og ERC benyttes primært når det er behov for børstecytologi eller terapeutiske prosedyrer som f.eks. blokking og stenting av strikturer. Klinisk bakgrunnsinformasjon er viktig for å utelukke aktuelle differensialdiagnoser som f.eks. sekundær scleroserende cholangitt som følge av langvarig steinsykdom.
Autoantistoffer kan være positive ved PSC, men spesifisiteten er lav. Leverbiopsi er ikke nødvendig for diagnosen, men kan være nyttig for å estimere grad av leverfibrose, og ved mistanke om overlapp mot AIH. Et klassisk biopsifunn ved PSC er såkalt «løkring-fibrose» omkring intrahepatiske galleveier (Figur 1C).
Hos pasienter med kjent IBD ses noen ganger bioptiske funn forenlig med PSC uten at det foreligger kaliberveksling i galleveiene. Dette defineres som «small duct PSC», en tilstand som har bedre prognose enn PSC generelt.
Oppfølging
Tidligere trodde man at ursodeoksykolsyre kunne bremse sykdomsprosessen, men nyere forskning har trukket dette i tvil, og preparatet brukes ikke lenger rutinemessig. Per i dag finnes ingen medisinsk behandling som demper sykdomsprosessen ved PSC. Det forventes at nye terapeutiske prinsipper (endre gallesyresammensetningen, manipulering av tarmflora, farnesoid X reseptor antagonisme, selektiv immunmodulering m.m.) kan komme til anvendelse i årene som kommer(12).
I fravær av effektiv medikamentell terapi må man i oppfølgingen av PSC-pasienter fokusere på forebygging og håndtering av komplikasjoner. Endokopisk behandling av gallegangsstrikturer kombinert med antibiotika når det er mistanke om ascenderende infeksjon står helt sentralt. Mange pasienter har stor nytte av kortvarig stent-behandling av galleveisstrikturer.
Overvåking i forhold til cholangiocarcinom er meget utfordrende. Den maligne utvikling kan komme snikende, og i tidlig stadium foreligger ofte ingen spesielle symptomer. Tumormarkøren CA 19-9 brukes, men hverken sensitivitet eller spesifisitet er tilfredsstillende. Man skal alltid tenke på cholangiocarcinom når det kliniske bildet forverres. Ved nytilkomne domi-nante gallegangstrikturer tas børstecytologi av det affiserte området, men sensitiviteten i forhold til cholangiocarcinom er relativt dårlig. Fortolkning av prøvematerialet er vanskelig og krever en erfaren cytopatolog. Diagnostikken må ofte suppleres med CT eller MR undersøkelse.
Prognose
I dag stilles PSC- diagnosen ofte på et relativt tidlig stadium, og sannsynligvis identifiseres flere pasienter med et mildere sykdomsforløp. Tidligere ble median transplantasjonsfri overlevelse angitt til ni år. En populasjonsbasert studie fra Nederland har vist median transplantasjonsfri overlevelse på 21 år (13). For mange er PSC likevel en svært alvorlig sykdom som etterhvert gir livstruende komplikasjoner. Levertransplantasjon er den eneste kurative behandling, og PSC er en av de vanligste årsaker til transplantasjon i Norge. Selv om sykdommen residiverer hos 20-30%, er resultatene gode med median 5-årsoverlevelse nær 90% (Figur 3).
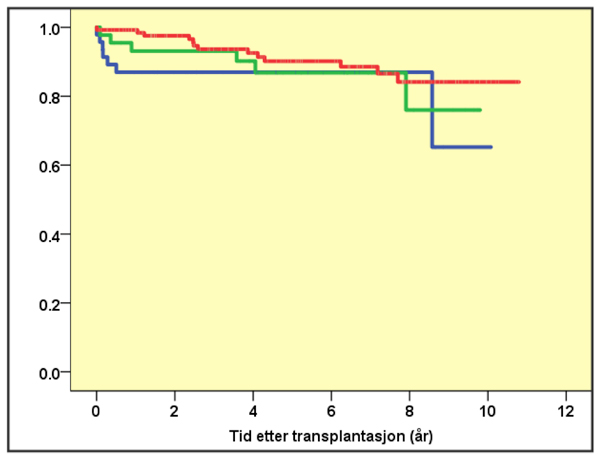
Figur 3. Overlevelse etter levertransplantasjon ved autoimmun hepatitt (blå linje), primær biliær cirrhose (grønn linje) og primær scleroserende cholangitt (rød linje) basert på data fra Nordisk levertransplantasjonsregister (www.scandiatransplant.org).
De autoimmune leversykdommene er heterogene, og det er et stort spekter fra de mildeste tilfeller til de med avansert organsykdom og livstruende komplikasjoner. Tidlig diagnose er viktig da medikamentell og endoskopisk terapi bedrer sykdomsforløpet hos mange. Alle tre autoimmune leversykdommer kan resultere i cirrhose. Optimal håndtering av cirrhose komplikasjoner er derfor avgjørende. En del av de sykeste pasientene gjennomgår levertransplantasjon med gode resultater.
Referanser



