Utskillelsen av kalsium og fosfat i nyren er underlagt en streng hormonell kontroll. Forstyrrelser i mineralmetabolismen utvikles tidlig hos pasienter med kronisk nyresvikt. I løpet av sykdomsutviklingen affiseres nær alle pasienter [2]. Forstyrrelsene ble lenge sett på som et direkte resultat av en gradvis reduksjon i mengden normalt fungerende nyrevev med en redusert aktivering av vitamin D til 1,25(OH)2 vitamin D med påfølgende redusert serum kalsium og fosfatretensjon. En kompensatorisk økning av PTH sikrer så at kalsium og fosfatverdiene blir holdt relativt stabile innenfor normalområdene inntil GFR faller under 20 ml/min/1,73 m2 (klassisk «trade off» hypotese) [3].
Parathyroideahormon (PTH)
PTH er den klassiske biomarkøren for SHPT og ved endestadium nyresvikt (end stage renal disease, ESRD) kan nivåene bli svært høye. På grunn av sin aktive rolle i benresorpsjon har PTH vært brukt som indikator på nyreassosiert bensykdom (adynamisk bensykdom og osteodystrofi/osteitis fibrosa cystica). Den direkte sammenhengen mellom PTH nivåer og histologiske forandringer i benvev har imidlertid vært svak. Ved nyresvikt akkumuleres en rekke ulike PTH fragmenter og en mulig årsak til inkonsistente funn, er bruk av forskjellige PTH assays. Tredje generasjons PTH assays måler det intakte hormonet bestående av aminosyre 1-84, men foreløpig har en ikke klart å vise at dette bedrer den diagnostiske treffsikkerheten [4].
Flere observasjonsstudier har vist en sammenheng mellom høy PTH og risiko for død og hjerte- og karsykdom, og PTH har i mange år blitt betraktet som et uremisk toksin med ødeleggende effekter på kardiomyocyttene. Den sterkeste lineære sammenhengen er funnet hos hemodialysepasienter der det ble påvist en signifikant økt risiko for hjerte- og karsykdom ved PTH verdier over 600 pg/ml [5]. Resultatene er imidlertid ikke entydige. I en annen studie av hemodialysepasienter var både høye og lave verdier av PTH assosiert med økt sykelighet og død, og det har blitt argumentert for at sammenhengen ikke er lineær, men U-formet [6]. Flere negative studier har blitt kritisert for å ha brukt statistiske metoder som kun ville påvise en lineær sammenheng om den fantes [7]. En ytterligere svakhet ved studiene er at dagens PTH assays ikke diskriminerer mellom oksiderte inaktive former for PTH og ikke-oksiderte former, og det kan se ut til at kun sistnevnte er assosiert med lavere dødelighet. Oksidativt stressnivå kan derfor vise seg å være en betydelig konfunderende faktor som det må tas høyde for i fremtidige studier av sammenhengen mellom PTH og risiko for uønskede hendelser [4].
SHPT med vekst av parathyroidea og økt utskillelse av PTH er en av hovedkomplikasjonene til kronisk nyresvikt. Det er et nært samspill mellom parathyroidea, ben og hjerte- og karsystemet og betegnelsen Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) ble i 2005 introdusert for å dekke et bredere klinisk syndrom bestående av elektrolyttforstyrrelser, bensykdom og kalsifisering av bløtvev og kartre [8]. Humane cellekulturer av glatte muskelceller (VSMC, vascular smooth muscle cells) differensierer mot osteoblastlignende celler når de utsettes for høye fosfatkonsentrasjoner. Kalsifiseringen av kartreet kan således sees som en aktiv prosess som har likheter med bennydannelse og understøtter oppfatningen av CKD-MBD som et klinisk syndrom [9].
Studier av pasienter med de arvelige fosfattapende sykdommene autosomal-dominant hypofosfatemisk rakitt (ADHR) og X-bundet hypofosfatemisk rakitt (XLH), samt tumor indusert osteomalaci (TIO), førte til identifikasjon av det fosfatregulerende hormonet fibroblast growth factor 23 (FGF23) i 2000 [10]. ADHR er forårsaket av en aktiverende FGF23 mutasjon, mens XLH pasienter har en rekke inaktiverende mutasjoner i et gen (PHEX) som styrer FGF23 ekspresjonen i ben og medfører en fenotype nær identisk med ADHR. αKlotho ble oppdaget i 1997 i forbindelse med museforsøk (random insertional mutation experiments) [11]. Mus uten FGF23 eller αKlotho utviste like fenotypiske trekk forbundet med det å eldes (vekst retardasjon, forkortet livslengde, osteomalaci, kalsifiseringer i bløtvev og kartre) og videre studier avdekket αKlotho som en obligat ko-receptor for FGF23.
Fylogenetiske studier av fibroblast growth factors (FGF) indikerer at de kan inndeles i 7 undergrupper og videre klassifiseres etter virkningsmekanisme som intrakrine (FGF11), auto/parakrine (FGF1,4,7,8 og 9) og endokrine (FGF19,21 og 23) [12]. De endokrine FGF er involvert i reguleringen av lipid-, glukose og mineralmetabolismen. FGF23 produseres i benvev og har sin viktigste virkning i nyren der den hemmer 1α-hydroksylasen (Cyp27b1) og derved aktiveringen av vitamin D og øker fosfatutskillelsen ved å internalisere ionekanaler (NaPi-2a og NaPi-2c kotransportere) i proksimale tubuli. FGF23 hemmer også syntesen og sekresjonen av PTH. Nettoeffekten av FGF23 er således reduserte fosfat (og kalsium) nivåer via samordnede virkninger på nyren og glandula parathyroidea [13].
αKlotho, med navn fra gresk mytologi, er uttrykt hovedsakelig i nyren og parathyroidea og sikrer slik FGF23s vevsspesifisitet som en obligat koreseptor for FGF23. αKlotho foreligger også i to sirkulerende isoformer og har FGF23 uavhengige effekter på kalsium (øker) og fosfat (senker) nivåene. Det gjenstår imidlertid mye forskning før αKlothos effekter er fullstendig kartlagt. Et hovedproblem har vært mangelfulle målemetoder, spesielt når det gjelder vevsbundet Klotho [14].
Reduksjonen i aktivt vitamin D, kommer tidlig ved kronisk nyresvikt, og står ikke i forhold til den reduserte nyremassen som først blir av betydning sent i sykdomsutviklingen. Oppdagelsen av FGF23 og påvisningen av økte nivåer ved kronisk nyresvikt har endret forståelsen av dette kompliserte samspillet radikalt. I tidlige stadier av nyresvikt er det er nær lineær sammenheng mellom nyrefunksjon (GFR) og FGF23 nivåene, mens denne sammenhengen blir eksponentiell i senere stadier der en kan se verdier opp mot 1000 ganger normalverdien. Årsaken til disse ekstreme verdiene er ikke fullstendig kartlagt, men kan sees på som et resultat av FGF23 resistens med redusert ekspresjon av αKlotho, figur 1 [13]. En tenker seg nå at fosfatverdien holdes innenfor ønsket nivå av FGF23 som virker direkte fosfaturisk på nyren og samtidig hemmer aktiveringen av vitamin D og derved også fosfat (og kalsium) absorpsjonen fra tarm og frigjøring fra benvev. Prisen vi betaler for å holde fosfatet normalt er høye verdier av både FGF23 (stiger tidlig) og PTH (stiger noe senere) [3].
![Figur 1 Foreslått tidsprofil for endringer i mineralstoffskiftet i relasjon til nyrefunksjon. Gjengitt fra Annual Review of Physiology 2013 [13]. Gjengitt med tillatelse.](https://indremedisineren.no/wp-content/uploads/2017/07/Bleskestad.-Figur-1.png)
Figur 1
Foreslått tidsprofil for endringer i mineralstoffskiftet i relasjon til nyrefunksjon.
Gjengitt fra Annual Review of Physiology 2013 [13]. Gjengitt med tillatelse.
Observasjonsstudier av en rekke ulike pasientpopulasjoner har funnet en sterk og uavhengig assosiasjon mellom FGF23 og hjerte- og karsykdom og død. FGF23 ser videre ut til også å ha en direkte skadelig effekt på kardiomyocyttene via en Klotho uavhengig virkning. Det er vist at FGFR4 oppreguleres i kardiomyocyttene ved høye FGF23 nivåer, og at FGF23 også virker via en calcineurinavhengig signalvei [15]. Det gjenstår å fastslå når beskyttelsesmekanismene mot forhøyet fosfat skyter over mål og gjør mer skade enn gavn.
I 2009 kom Kidney Disease Improving Global Outcomes (KDIGO) behandlingsretningslinjene for CKD-MBD som fortsatt er gjeldende [16]. Godkjent behandling av forstyrrelser i mineralmetabolismen består i dag av fosfatbindere (fosfatreduksjon) og aktive vitamin D analoger og cinacalcet (PTH reduksjon). Aktive vitamin D analoger senker PTH, men øker samtidig kalsium og fosfat, noe som begrenser bruken. Cinacalcet er et peroralt «kalsimimetikum» som
ved binding til den kalsiumfølsomme reseptoren (Calcium Sensing Receptor (CaSR)) i parathyroidea hemmer PTH sekresjonen og indirekte senker fosfat og FGF23 nivåene uten samtidig å gi pasienten en positiv kalsiumbalanse. Det har av denne grunn vært knyttet sterke forhåpninger til denne medikamentgruppen.
The Evaluation of Cinacalcet Hydrochloride Therapy to Lower Cardiovascular Events (EVOLVE) studien er den første store randomiserte, kontrollerte intervensjonsstudien som har vurdert om en senkning av PTH ville føre til en reduksjon i hjerte‐ og karsykdom og død hos hemodialysepasienter [17]. Studien omfattet 3883 pasienter med ESRD og SHPT der intervensjonsgruppen ble gitt cinacalcet og kontrollgruppen fikk standard behandling. Hjerte‐ og karsykdom omfattet i denne studien hjerteinfarkt, hospitalisering for ustabil angina pectoris, hjertesvikt eller perifer karsykdom inkludert revaskulariseringsinngrep på underekstremitetene og ikke traumatisk amputasjon. Begrunnelsen for det sammensatte endepunktet var at studiegruppen på forhånd ikke visste hvilken type karsykdom de ville lykkes i å redusere. Studien ble analysert etter ”Intention To Treat” (ITT) prinsippet og var i så måte negativ. Et av problemene var blant annet at de som et resultat av randomiseringen fikk en intervensjonsgruppe som var 1 år eldre enn kontrollgruppen. En stor andel av pasientene sluttet å ta medikamentet (mange «drop-outs»), og det var også en stor andel ”drop ins”, d.v.s. at pasientene i placebogruppen fikk Cinacalcet, som er kommersielt tilgjengelig. Dette reduserte den statistiske styrken i studien.
I årets januarutgave av JAMA [18, 19] er det publisert tre studier med et annen-generasjons intravenøst kalsimimetikum, etelcalcetide, som kan gis tre ganger per uke sammenfallende med dialysebehandling. Studiedesignet hindret problemene med «drop-outs» og «drop-ins», og slik ble forskjellen mellom behandlings- og kontrollgruppen opprettholdt gjennom studieperiodene. I de to placebokontrollerte studiene over 27 uker oppnådde behandlingsgruppene det primære endepunktet (30 % reduksjon i PTH) i 74,0 og 75,3 % v.s. 8,3 og 9,6 % i kontrollgruppene, p<0.001 for begge studiene [19]. Etelcalcetide reduserte signifikant serum fosfat og FGF23 nivåene. Det foreligger imidlertid ingen studier med harde endepunkter.
Det primære målet med behandling av CKD-MBD er å redusere de toksiske virkningene av forhøyet serum fosfat. Mye tyder på at behandlingen bør initieres tidligere enn det dagens behandlingsretningslinjer fra 2009 anbefaler. Harde endepunktsstudier som inkluderer pasienter i CKD stadium 3 og 4 før karsykdom er etablert og videre studier av pasienter i hemodialyse er nødvendige for evalueringen av fosfatreduserende behandling sin effekt på hjerte- og karsykdom.
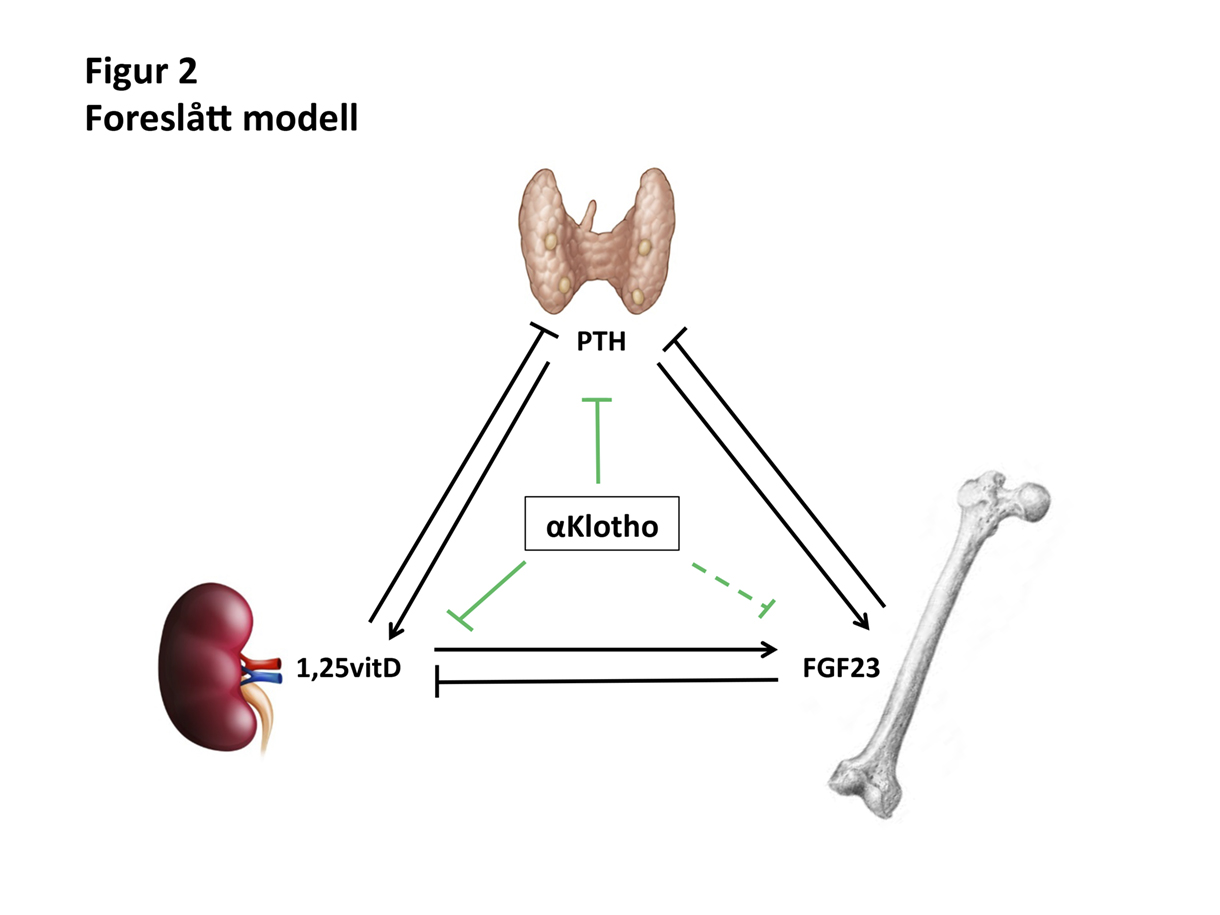
Figur 2
Foreslått modell: Det foreligger klassiske endokrine feedback sløyfer mellom vitamin D, FGF23 og PTH. Sirkulerende αKlotho (sentral boks i figuren) stammer under normale forhold i all hovedsak fra nyren og det er holdepunkter for at αKlotho har en sentral plass i reguleringen av mineralmetabolismen (jf ref).
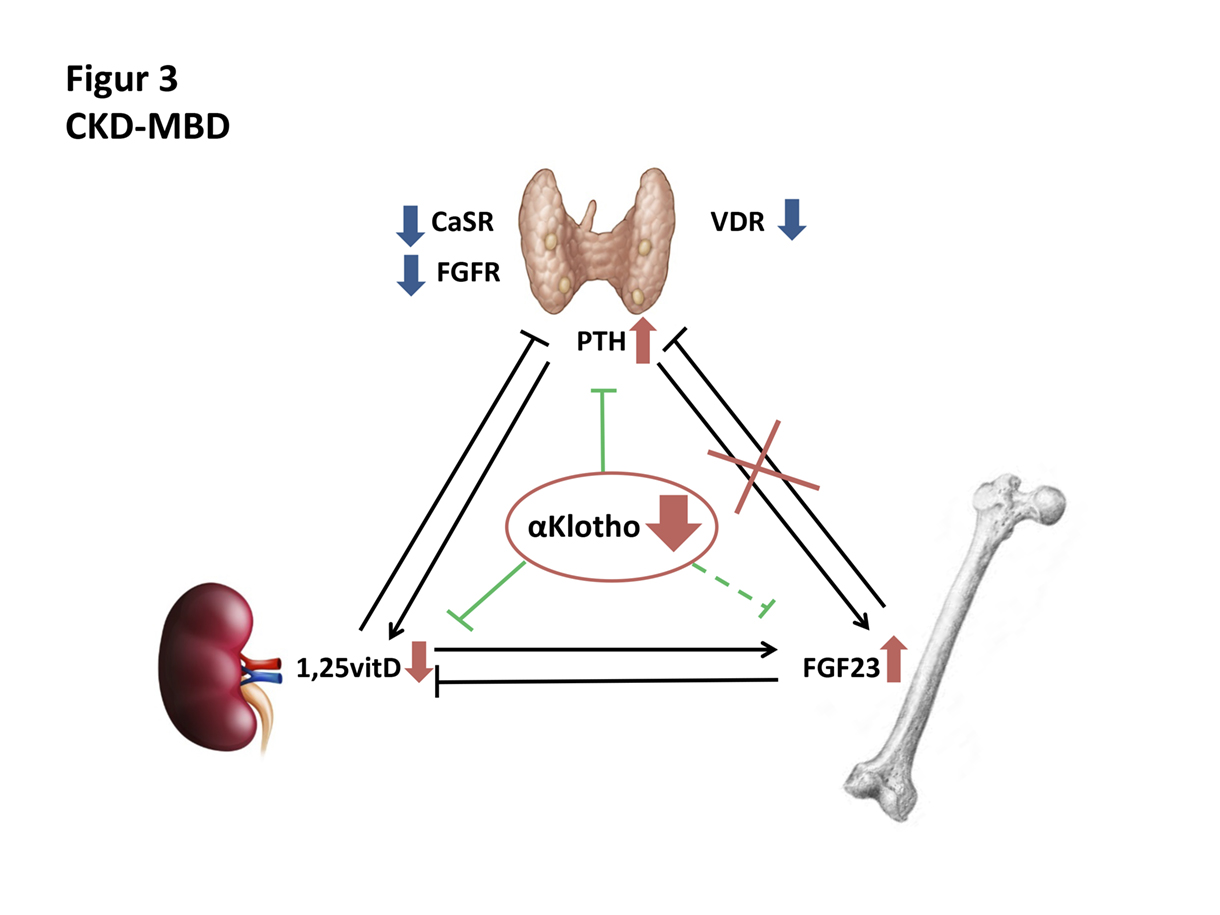
Figur 3
CKD-MBD: Hva som er utløsende faktor ved kronisk nyresvikt er ikke kartlagt, men det ser ut for at αKlotho reduseres før FGF23 stiger. Nyren klarer ikke å opprettholde produksjonen av αKlotho og aktivt vitamin D. For å holde serum fosfat- og serum kalsiumnivåene innenfor normalområdet, stimuleres benvev til FGF23 produksjon og parathyroidea til PTH produksjon. Ved utvikling av SHPT reduseres kalsium sensing reseptor- (CaSR), FGFreseptor- (FGFR) og vitamin D reseptor- (VDR) tettheten i den hyperplastiske parathyroideakjertelen og FGF23 klarer ikke å hemme PTH produksjonen. Det utvikles en ende-organ hormonresistenstilstand.
Ref: Neyra JA, HU MC. Potential application of Klotho in human CKD. Bone 2017. In press
Referanser
1. Hajhosseiny R, Khavandi K, Goldsmith DJ. Cardiovascular disease in chronic kidney disease: untying the Gordian knot. Int J Clin Pract 2013;67(1):14-31
2. Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007;71(1):31-38
3. Gutierrez OM. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: updating the «trade-off» hypothesis. Clin J Am Soc Nephrol 2010;5(9):1710-1716
4. Mazzaferro S, Tartaglione L, Rotondi S, et al. News on biomarkers in CKD-MBD. Semin Nephrol 2014;34(6):598-611
5. Block GA, Klassen PS, Lazarus JM, et al. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004;15(8):2208-2218
6. Floege J, Kim J, Ireland E, et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol Dial Transplant 2011;26(6):1948-1955
7. Natoli JL, Boer R, Nathanson BH, et al. Is there an association between elevated or low serum levels of phosphorus, parathyroid hormone, and calcium and mortality in patients with end stage renal disease? A meta-analysis. BMC Nephrol 2013;14:88
8. Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006;69(11):1945-1953
9. London GM. Mechanisms of arterial calcifications and consequences for cardiovascular function. Kidney International Supplements 2013;3:442-445
10. Consortium A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000;26(3):345-348
11. Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997;390(6655):45-51
12. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem 2011;149(2):121-130
13. Hu MC, Shiizaki K, Kuro-o M, et al. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 2013;75:503-533
14. Olauson H, Vervloet MG, Cozzolino M, et al. New insights into the FGF23-Klotho axis. Semin Nephrol 2014;34(6):586-597
15. Leifheit-Nestler M, Grosse Siemer R, Flasbart K, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant 2016;31(7):1088-1099
16. Kidney Disease: Improving Global Outcomes CKDMBDWG. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl 2009(113):S1-130
17. Investigators ET, Chertow GM, Block GA, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med 2012;367(26):2482-2494
18. Block GA, Bushinsky DA, Cheng S, et al. Effect of Etelcalcetide vs Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: A Randomized Clinical Trial. JAMA 2017;317(2):156-164
19. Block GA, Bushinsky DA, Cunningham J, et al. Effect of Etelcalcetide vs Placebo on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: Two Randomized Clinical Trials. JAMA 2017;317(2):146-155



