Sigbjørn Berentsen, overlege, seniorforsker, dr. med., Seksjon for forskning og innovasjon Haugesund sjukehus, Tor Hervig, seksjonsoverlege, professor, dr. med., Eining for immunologi og transfusjonsmedisin, Haugesund sjukehus og Klinisk institutt 2, Universitetet i Bergen, Geir E. Tjønnfjord, avdelingsleder, professor, dr. med., Avdeling for blodsykdommer, Oslo universitetssykehus og KG Jebsen senter for B-cellemaligniteter, Institutt for klinisk medisin Universitetet i Oslo Korrespondanse: Sigbjørn Berentsen, sigbjorn.berentsen@haugnett.no
Autoimmun hemolytisk anemi defineres som økt destruksjon av erytrocytter forårsaket av autoantistoff mot antigener på celleoverflaten. Sykdomsgruppen er uvanlig, men leger ved de fleste medisinske sykehusavdelinger vil møte slike pasienter. Emnet er også viktig innen transfusjonsmedisin.
En norsk oversiktsartikkel ble publisert i 2009 (1). Siden har det tilkommet nye funn vedrørende etiologi, patogenese, diagnostikk og behandling (2-4). Tidligere har klinisk praksis i stor grad vært basert på ekspertanbefalinger, kasuistikker og erfaring, men de siste par tiårene er det publisert flere prospektive og andre systematiske kliniske studier. Både i Norge og internasjonalt ser man eksempler på at utredning og behandling kan forbedres (2, 5, 6). I 2020 ble det for første gang publisert et internasjonalt konsensusarbeid om diagnostikk og behandling (2), og det finnes også britiske retningslinjer (3, 7). Denne oversikten bygger på et systematisk søk i PubMed, det internasjonale konsensusdokumentet og egen forskning. Autoimmun hemolytisk anemi er en samlebetegnelse for flere tilstander som har ulik etiologi, patogenese og klinisk sykdomsbilde, og som behandles ulikt (Tabell 1).
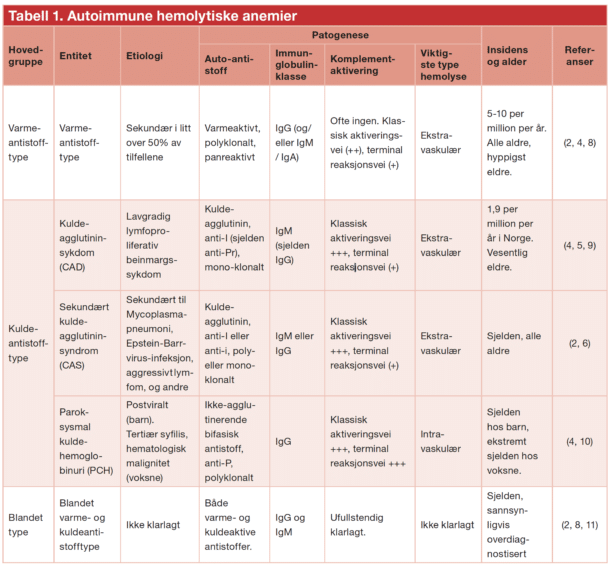
Et varmeantistoff har størst affinitet til antigenet ved 37oC. Ved varmeantistoffmediert hemolyse er autoantistoffene polyklonale, dvs. produsert av ikke-klonale B- lymfocytter (2). De er vanligvis av IgG-type, men kan være IgM eller en sjelden gang IgA. Patogenesen er sammensatt (Figur 1). T-cellenes regulering av B-cellesystemet spiller også en sentral rolle. Signalsubstansen cytotoksisk T-lymfocyttassosiert protein 4 (CTLA-4) aktiverer regulatoriske T-celler (Treg-celler), som er viktige for immunologisk toleranse. Polymorfisme av genet for CTLA-4 kan disponere for autoimmune sykdommer, ikke minst autoimmune cytopenier. Også den cellulære signalveien for «programmert celledød 1» (PD-1) er et sjekkpunkt for immunologisk toleranse, og medikamentell hemming av PD-1 medfører økt risiko for autoimmunitet (12).
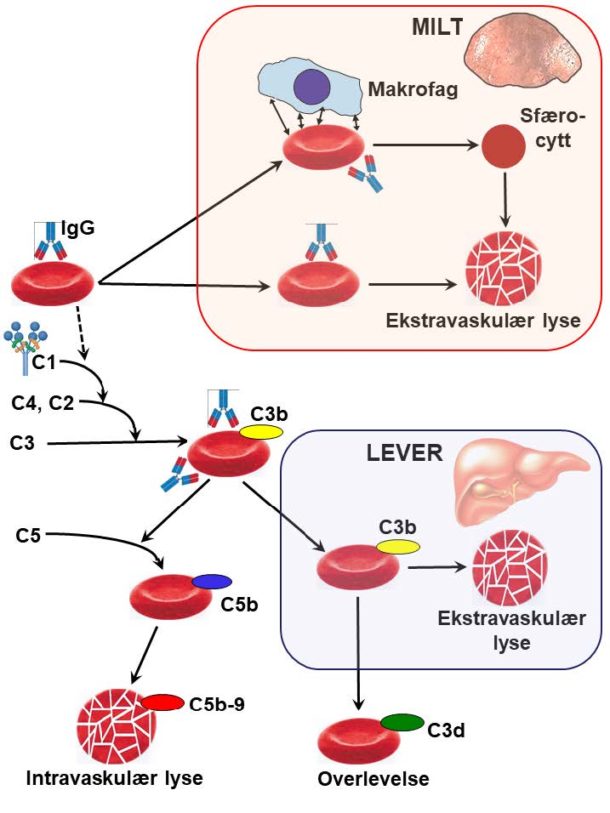
Hos nær 50% av pasientene med varmeantistoffmediert hemolyse påvises det ingen tilgrunnliggende eller assosiert sykdom, og tilstanden kalles da primær (2). Litt over halvparten av tilfellene er sekundære, dvs. assosiert med kronisk lymfatisk leukemi, systemisk lupus erythematosus (SLE), vanlig variabel immunsvikt eller andre immunologiske eller lymfoproliferative sykdommer (8). Samtidig eller sekvensiell kombinasjon med immun trombocytopeni, kalles Evans’ syndrom. Iblant kan den lymfoproliferative sykdommen først erkjennes flere år etter debut av hemolyse. Autoimmun hemolytisk anemi medfører økt risiko for venøs tromboembolisme (2, 5, 13). «
Kuldeantistoffer har størst affinitet til antigenet ved 0-4oC, men kan reagere også ved høyere temperatur (4). Kuldeantistoffer som agglutinerer erytrocytter, kalles kuldeagglutininer. Den høyeste temperaturen der agglutinasjon kan påvises, kalles den termale amplituden, og kuldeagglutininer med en høyere termal amplitude enn ca. 28oC vil være patogene (10, 28). Kuldeagglutinintiteret er den inverse verdien av den største serum- eller plasmafortynningen der agglutinering av erytrocytter kan påvises ved en gitt temperatur. Dersom for eksempel erytrocytter agglutinerer ved 4oC når pasientplasma tilsettes i fortynning 1:64, men ikke ved fortynning 1:128, angis titeret som 64 ved 4oC.
Kuldeagglutininsykdom (cold agglutinin disease, CAD) defineres som en autoimmun hemolytisk anemi der autoantistoffet er et kuldeagglutinin og der det ikke kan påvises noen tilgrunnliggende klinisk sykdom (2, 6). Flere studier har vist at slike pasienter, som tidligere gjerne fikk diagnosen primær eller idiopatisk kuldeagglutininsykdom, har en klonal, lymfoproliferativ beinmargssykdom som kan være vanskelig å diagnostisere (5, 9). Slike beinmargsfunn har i praksis ofte blitt klassifisert som lymfoplasmacytisk lymfom eller splenisk marginalsonelymfom, men nyere studier har vist at det i de aller fleste tilfeller dreier seg om en relativt ensartet histopatologisk tilstand som har fått betegnelsen kuldeagglutinin-assosiert lymfoproliferativ beinmargssykdom (9).
Kuldeagglutininet er monoklonalt, vanligvis et IgM, og produseres av klonale lymfocytter i beinmargen. Når kuldeagglutininet bindes til sitt antigen, initieres komplementmediert hemolyse som vist i Figur 2. Avkjøling i de kaldere delene av blodsirkulasjonen fører til agglutinering av erytrocytter. Dette vanskeliggjør passasje i kapillærene, og 50-60% av pasientene har kuldeinduserte sirkulasjonssymptomer som kan variere fra lett akrocyanose til invalidiserende Raynaud-liknende fenomener (5).
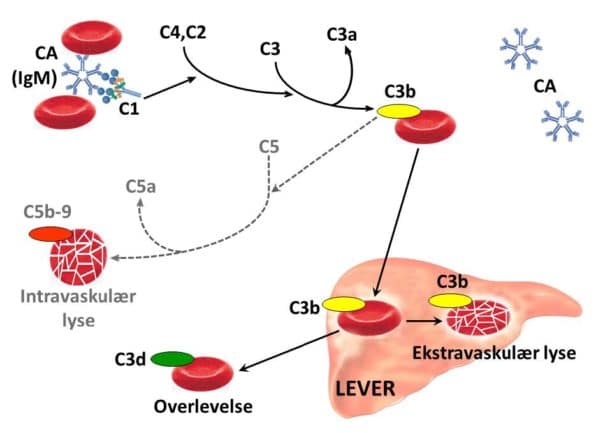
Noen personer får tilfeldig påvist polyklonale kuldeagglutininer i lavt titer, med lav termal amplitude og uten hemolyse eller kliniske symptomer. De har ikke kuldeagglutininsykdom eller -syndrom (6).
Sekundært kuldeagglutininsyndrom Betegnelsene kuldeagglutininsykdom og kuldeagglutininsyndrom ble tidligere brukt synonymt. I dag skiller vi mellom kuldeagglutininsykdom (en veldefinert klinisk-patologisk entitet som forklart ovenfor) og sekundært kuldeagglutininsyndrom (cold agglutinin syndrome, CAS) (2, 6). Sistnevnte er en kuldeagglutininmediert hemolytisk anemi som oppstår sekundært til annen klinisk sykdom. De fleste tilfeller inntrer under eller etter infeksjon med Mycoplasma pneumoniae (14). Andre infeksiøse årsaker er Epstein- Barr-virus, cytomegalovirus, andre virus eller Chlamydia pneumoniae. Syndromet kan også være assosiert med malign sykdom, oftest aggressivt B-cellelymfom (6).
Paroksysmal kuldehemoglobinuri Ved paroksysmal kuldehemoglobinuri er autoantistoffet bifasisk og kalles Donath-Landsteiners antistoff (2, 10). Det bindes til sitt antigen ved temperaturer under sentral kroppstemperatur, men påfølgende komplementaktivering foregår etter oppvarming til 37oC i den sentrale sirkulasjonen. Sykdommen forekommer nå nesten bare som en sjelden, postviral komplikasjon hos små barn (10, 15).
Medikamentassosiert hemolytisk anemi De fleste medikamentbetingete hemolytiske anemier medieres via immunologiske mekanismer (7). Over 100 legemidler er rapportert å kunne gi denne bivirkningen, som likevel forekommer sjelden. Tidligere var årsaken som regel alfametyldopa eller store penicillindoser, men i dag dominerer ceftriaxon, andre kefalosporiner og ikke-steroide antiinflammatoriske midler (2, 7). Ved kronisk lymfatisk leukemi kan behandling med fludarabin eller klorambucil, særlig i monoterapi, øke risikoen for autoimmun hemolyse (2). Ikke uventet kan også sjekkpunkthemmere som brukes i immunologisk kreftbehandling, særlig PD-1-inhibitorer, gi autoimmune komplikasjoner, bl.a. hemolytisk anemi (12).
Blandet varme- og kuldeantistofftype Insidensen av denne undergruppen er omdiskutert, men sannsynligvis er den svært sjelden og overdiagnostisert (11). Patogenesen er ufullstendig klarlagt og antakelig heterogen.
Figur 3 viser en utredningsalgoritme, og Tabell 2 gir en oversikt over aktuelle prøver. Ved hemolyse finner man vanligvis forhøyede nivåer av laktatdehydrogenase og bilirubin, mens haptoglobin er nedsatt eller ikke detekterbart. Retikulocytose støtter hemolysediagnosen. Hos noen pasienter har imidlertid autoantistoffet også aktivitet mot erytrocyttforstadier i beinmargen, og derfor kan retikulocyttallet enkelte ganger være normalt eller lavt (2, 8). Når hemolyse er påvist, utfører man direkte antiglobulintest (DAT) for å påvise autoimmun patogenese. Positiv test viser at det finnes immunglobulin og/eller komplement på erytrocyttoverflaten.
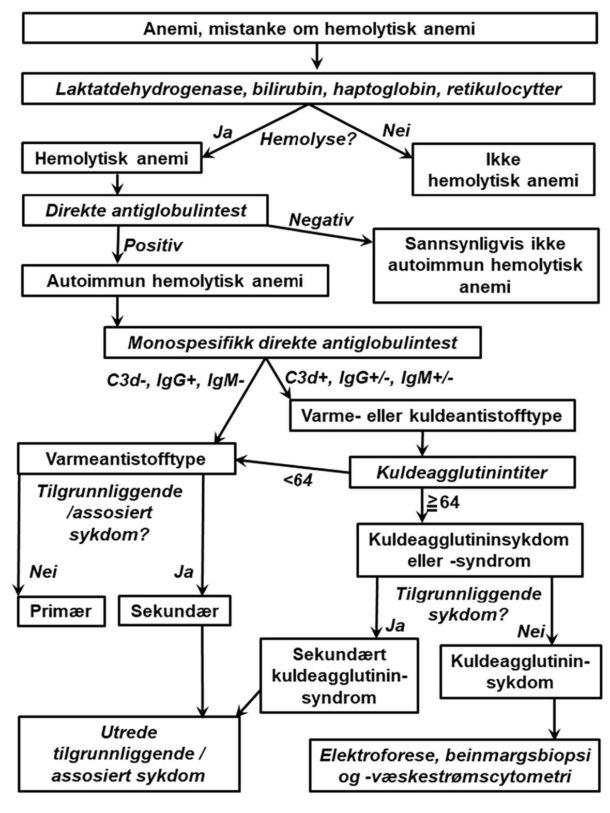
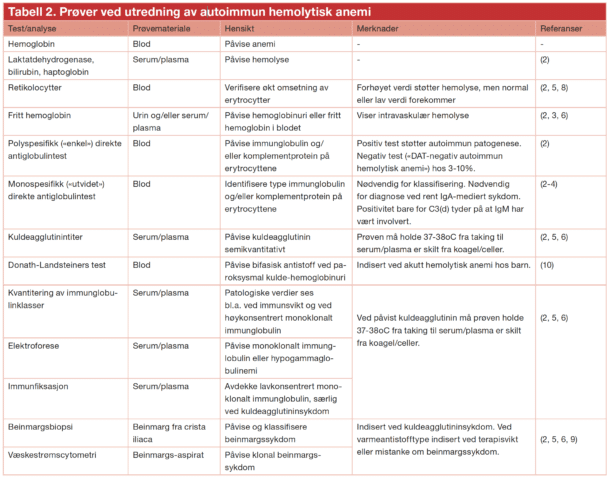
Ved positiv DAT må man alltid utføre monospesifikk («utvidet») DAT (2). Denne testen undersøker hvilken immunglobulinklasse eller hvilket komplementprotein som er tilstede på celleoverflaten. Ved varmeantistofftypen er dette oftest IgG, men det kan være IgA i tillegg eller en sjelden gang alene (2, 8). Komplement, oftest i form av C3d, påvises i omlag halvparten av tilfellene og viser komplementaktivering (Figur 1). Dersom varmeaktivt IgM er involvert i patogenesen, kan dette løsne fra celleoverflaten før det kan påvises i laboratoriet, men i mellomtiden har det aktivert komplement (16). DAT som er positiv for C3d og negativ for IgG, er derfor et tegn på at IgM har vært involvert (2, 16).
Autoimmun hemolytisk anemi med negativ DAT forekommer i 3-10% av tilfellene og er en vanskelig eksklusjonsdiagnose der spesielle væskestrømscytometri- eller elueringsteknikker enkelte ganger kan være til hjelp (2). Sistnevnte innebærer at man eluerer eventuelt immunglobulin fra erytrocyttoverflaten og deretter utfører indirekte antiglobulintest (IAT) i eluatet. Hos noen slike pasienter kan man finne tegn til klonal T-lymfocyttsykdom. Ved rent IgA-mediert hemolyse av varmeantistofftype vil polyspesifikk («enkel») DAT være negativ, men disse sjeldne tilfellene vil oppdages om man likevel utfører monospesifikk test.
Ved kuldeagglutininsykdom eller -syndrom er monospesifikk DAT alltid sterkt positiv for C3d og vanligvis negativ for immunglobuliner, men kan være svakt positiv for IgG (5, 6). Dette er ikke nok til å stille diagnosen. Man må også undersøke kuldeagglutinintiter, som vil være minst 64, oftest mye høyere. Det må tas spesielle hensyn ved prøvetakingen som vist i Tabell 2. Paroksysmal kuldehemoglobinuri bør vurderes hos barn med akutt hemolytisk anemi, og mistanken må avklares ved å teste for bifasisk autoantistoff med Donath-Landsteiners test (10).
Som i annen diagnostikk er sykehistorie og klinikk viktig (2, 6). Bruker pasienten medikamenter? Har hun/ han kjent lymfoproliferativ eller autoimmun sykdom? Er lymfeknuter eller milt forstørret? Er det kliniske tegn til kuldeagglutininsykdom?
Ved varmeantistofftypen må man deretter ut fra klinikk og hematologiske og immunologiske markører vurdere om pasienten kan ha en sykdom som er assosiert med sekundær autoimmun hemolyse (2). Serumelektroforese med immunglobulinkvantitering bør gjøres ved alle undertyper, bl.a. fordi vanlig variabel immunsvikt, hvor hypogammaglobulinemi er et sentralt element, kan debutere med autoimmune cytopenier. Ved kuldeagglutininsykdom må årsaker til sekundært kuldeagglutininsyndrom utelukkes (6, 14). Den lymfoproliferative beinmargsaffeksjonen ved kuldeagglutininsykdom bør verifiseres ved biopsi og væskestrømscytometri, men i et mindretall av tilfellene greier man ikke å påvise den (5, 9). En stor, deskriptiv studie har vist at vellykket påvisning av kuldeagglutininassosiert lymfoproliferativ beinmargssykdom oppnås oftere når biopsiene blir undersøkt sentralt ved en patologiavdeling med erfaring i å gjenkjenne denne entiteten (5). Også ved varmantistofftypen bør biopsi og væskestrømscytometri av beinmarg utføres på liberalt grunnlag, iallfall ved terapisvikt eller mistanke om beinmargssykdom.
Autoimmun hemolytisk anemi er en samlebetegnelse for flere tilstander som har ulik etiologi, patogenese og klinisk sykdomsbilde, og som behandles ulikt.
Varmeantistofftype
Førstelinjebehandling er prednisolon 60-100 mg eller 1 mg/kg kroppsvekt per dag i 2-3 uker med påfølgende langsom nedtrapping ved respons eller seponering ved terapisvikt (2, 3). Dersom pasienten etter 3-6 måneder har tålt nedtrapping til 7,5-10 mg daglig uten residiv av behandlingstrengende anemi, forsøkes gradvis full seponering. Rundt 80% responderer, men bare 30-40% er i vedvarende remisjon etter et år (2, 17).
Tillegg av rituksimab i første linje (375 mg/m2 fire ganger med én ukes intervall eller 1000 mg fast dose to ganger med to ukers intervall) er vist å øke antall langvarige remisjoner og bør overveies ved alvorlig sykdom (2, 17, 18). «Alvorlig» betyr først og fremst hemoglobinnivå under 8 g/dL, men også IgA-mediert, blandet eller DAT-negativ autoimmun hemolytisk anemi er vist å ha et alvorligere forløp (2, 8).
Anbefalt annenlinjebehandling er i dag rituksimab (2). Responsraten er 70-80%, men 30% får residiv innen tre år. Splenektomi anbefales først ved terapisvikt på rituksimab (2, 3). Rundt 70% responderer, men ulempene er risiko for alvorlig bakteriell infeksjonssykdom og ytterligere økt tromboserisiko.
Behandlingsvalg i seinere linjer bygger delvis på sammenslåtte («pooled») kasuistikker. Disse bør tolkes med skepsis fordi de vil være påvirket av seleksjon, publikasjonsskjevhet og heterogene responskriterier (2, 3). Azatioprin, syklofosfamid, ciklosporin, bortezomib og andre immunsuppressiver kan ha effekt. Lavdosert langtidsbehandling med prednisolon er et alternativ hos pasienter som hadde effekt av kortikosteroider initialt (2). Inklusjon i prosepktive studier bør foretrekkes framfor lite evidensbasert behandling utenfor studier. Studier pågår med inhibitorer av den neonatale Fc-reseptoren, fostamatinib (en splenisk tyrosinkinase-hemmer) og parsaclisib (en PI3Kδ-inhibitor med effekt mot B-lymfocytter).
Ved sekundær varmeantistoffmediert hemolytisk anemi er behandling av grunnsykdommen indisert dersom denne er behandlingstrengende i seg selv (2, 7). Ved terapisvikt på kortikosteroider bør man overveie å behandle grunnsykdommen selv når den ellers ikke oppfattes som behandlingstrengende.
Den internasjonale konsensusgruppen anbefaler venøs tromboseprofylakse ved markert hemolyse dersom pasienten er hospitalisert eller har andre risikofaktorer i tillegg. Osteoporoseprofylakse bør overveies ved behandling med prednisolon >7,5 mg/døgn i 3 måneder eller mer, og profylakse mot Pneumocystis jirovecii ved prednisolondoser >20 mg/døgn i én måned (2).
Kuldeagglutininsykdom
Kuldeagglutininsykdom skal ikke behandles med kortikosteroider. De har sjelden effekt, og stort sett bare i uakseptabelt høye doser (3, 5). Pasienter med lett anemi trenger ingen medikamentell behandling unntatt ved plagsomme sirkulasjonssymptomer eller uttalt fatigue (2, 5). Ikke-medikamentelle tiltak omfatter beskyttelse mot kulde. I sykehus bør pasienten holdes varm og kalde infusjonsvæsker bør unngås (2, 6).
Behandling rettet mot den patogene B-celleklonen har effekt ifølge flere prospektive studier. Rituksimab monoterapi induserer partiell remisjon hos 50-60%, men et flertall residiverer innen 12-15 måneder (5, 19). I en studie der pasientene fikk tillegg av peroralt fludarabin, observerte vi mye høyere responsrater og lengre responsvarighet, men også mer akutt og sein toksisitet (5, 20). Kombinasjonsbehandling med rituksimab og bendamustin i fire sykluser er vist å gi 78% responsrate, 53% komplett respons og median estimert responsvarighet på mer enn 88 måneder, men ofte lang tid til respons (5, 21). Bortezomib monoterapi har vist effekt, men bare 32% responsrate (2, 4).
Komplementhemmende behandling er lovende, men virker ikke på sirkulasjonssymptomene (4, 22). Effekt av sutimlimab, et monoklonalt antistoff mot komplementprotein C1s, er nå dokumentert i en fase 3-studie (23). Også peptidet pegcetacoplan, en C3-hemmer, ser i foreløpige studier ut til å ha effekt.
Vi anbefaler rituksimab pluss bendamustin i førstelinjesituasjonen til behandlingstrengende pasienter uten vesentlig komorbiditet, ellers rituksimab monoterapi (22). Inklusjon i en prospektiv studie kan være et godt alternativ.
Indikasjonene må være restriktive, men transfusjon må ikke unnlates ved livstruende anemi.
Andre typer
Sekundært kuldeagglutininsyndrom behandles ved å behandle grunnsykdommen om mulig (6, 7, 14). Ved paroksysmal kuldehemoglobinuri finnes ingen etablert medikamentell terapi, men det er nylig rapportert rask og dramatisk bedring av den postvirale formen hos et barn etter en enkelt dose av komplement-C5- hemmeren eculizumab (15). Medikamentassosiert hemolytisk anemi behandles med seponering av mistenkte medikamenter, men ved visse undertyper inntrer bedringen først lenge etter seponering. Initialbehandling med kortikosteroider kan forsøkes i mellomtiden, men dokumentasjonen er usikker (2, 7).
Transfusjon
Transfusjon ved varmeantistoffmediert immunhemolytisk anemi krever særskilte hensyn, da pasienten danner panreaktive antistoffer som binder seg til både autologe og allogene erytrocytter (24-26). Screening for irregulære antistoffer vil være positiv, og utvidet forlikelighetsprøve vil nesten alltid vise inkompatibilitet. Ofte vil det være umulig å identifisere eventuelt alloantistoff fordi autoantistoffet er svært sterkt. Det vil være risiko for akutte transfusjonsreaksjoner og dannelse av nye alloantistoffer, som kan maskeres av autoantistoffene og gi økende transfusjonsproblemer.
Indikasjonene må være restriktive, men transfusjon må ikke unnlates ved livstruende anemi. Transfusjon er først og fremst indisert ved svært raskt hemoglobinfall eller ved sirkulatoriske, kardiale eller cerebrale manifestasjoner av anemien, ikke ved et bestemt hemoglobinnivå (2, 3). Transfusjon bør utføres i nært samarbeid med blodbank og helst ved en spesialavdeling.
Man må akseptere at blodet ikke er kompatibelt ved testing. Om tiden tillater bør det brukes fenotypelike erytrocytter, som forebygger danning av alloantistoffer (24, 25). Typingen må inkludere Rh-, Kell-, MNS-, Kidd- og Duffy-antigener. Transfusjonen innledes med en biologisk forlikelighetstest, som innebærer at man først infunderer ca. 20 mL erytrocyttkonsentrat raskt, venter 20 minutter, og fortsetter om det ikke er tegn til transfusjonsreaksjon (2, 24).
Ved kuldeagglutininsykdom gjelder andre forholdsregler. Ofte finner man forlikelig blod, men avkjøling in vivo ved kald infusjon kan medføre agglutinering og økt hemolyse. Pasienten og ekstremiteten som velges for transfusjon må holdes varm, og bruk av blodvarmer anbefales (2, 3, 6).
Autoimmun hemolytisk anemi er en heterogen gruppe sykdommer som behandles ulikt. Presis diagnose av undertype og eventuell assosiert eller tilgrunnliggende tilstand må tilstrebes. Kortikosteroider er førstelinjebehandling ved varmeantistofftypen, men er ikke indisert ved kuldeagglutininsykdom. Behandling ved kuldeagglutininsykdom rettes mot den patogene Bcelleklonen. Når første- og eventuell annenlinjebehandling svikter, bør inklusjon i en prospektiv studie overveies framfor dårlig dokumentert tredjelinjebehandling. Transfusjon krever spesielle forholdsregler avhengig av sykdomstype.



