En mann i 80-årene utskrives fra sykehus etter å ha vært innlagt med atraumatisk hevelse og smerte i høyre ben. Tre uker senere utvikler han spontane hud -og muskelblødninger. Hevelsen i høyre ben viste seg å ha en langt sjeldnere årsak enn først antatt.
Daniel Kvarven, LIS, Bærum sykehus
Torkild Høieggen Pedersen, overlege, Bærum sykehus
I et travelt akuttmottak innlegges en eldre mann med funksjonssvikt og smerter i høyre arm og høyre ben. Mannen er i begynnelsen av 80-årene og har til vanlig et godt funksjonsnivå uten behov for kommunal bistand. Han er under behandling med Albyl-E, dipyramidol og statin etter et tidligere transitorisk iskemisk attakk (TIA) og implantasjon av biologisk aortaklaff (TAVI). Pasienten er ellers somatisk frisk og kognitivt velfungerende for sin alder.
Tre uker tidligere hadde mannen vært innlagt ved medisinsk avdeling med atraumatisk hevelse og smerter i høyre legg. Under innleggelsen på totalt fem døgn ble han bredt utredet med blodprøver, ultralyd og skjelettrøntgen uten at man fant tegn til hverken infeksjon, blodpropp eller skjelettskade. Det ble konkludert med at plagene trolig skyldtes overbelastning eller et atypisk urinsyregiktanfall. Han ble deretter utskrevet med en kortvarig prednisolon-kur og informert om å ta kontakt ved forverring.
Han hadde nå tatt kontakt igjen grunnet betydelig smerteforverring og funksjonssvikt. De siste 1-2 ukene før innleggelsen hadde smerten og hevelsen i høyre legg spredt seg til hele høyre underekstremitet og det hadde tilkommet smerter i høyre arm og på høyre side av brystveggen. Han klarte ikke lenger sove om natta på grunn av sterke smerter og var ute av stand til å utføre basale daglige gjøremål. Utover smerter og funksjonstap hadde pasienten ikke merket andre symptomer.
Ved undersøkelse hadde pasienten diffus hevelse av høyre lår- og leggmuskulatur samt store ekkymoser over høyre hemithorax og høyre arm. Det var god bevegelse over ledd, men lår- og leggmuskulaturen var stiv, spent og svært palpasjonsøm. Blodprøvene ved innkomst viste et lite hemoglobinfall fra forrige innleggelse på 10.6-9.9 g/dl (13.4-17.0), kreatinkinase 608 U/L (<280), CRP 33 mg/l (<5) og SR 43 mm/t (<19), ellers vesentlig normale blodprøver, inkludert trombocyttantall.
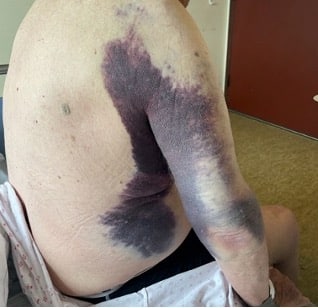
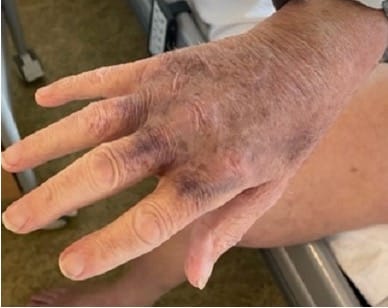
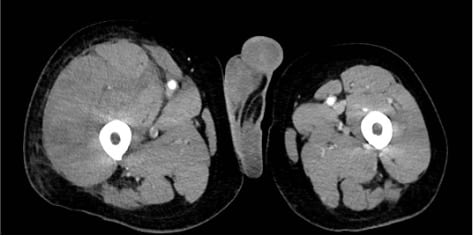
På bakgrunn av atraumatiske hudblødninger uten åpenbar årsak ble det rekvirert et bredt panel av blodprøver, inkludert INR, aktivert partiell tromboplastintid (APTT) og fibrinogen. Det ble tatt CT thorax, abdomen, bekken og høyre lår. Pasienten ble deretter innlagt på medisinsk avdeling for videre utredning. Noen timer senere kom svaret på de siste blodprøvene som viste et ytterligere hemoglobinfall fra 9.9-8.3 g/dl, INR 1.1 (0.9-1.2), APTT 60s (22-34 sekunder) og fibrinogen 4.9 g/L (2.0-4.1 g/L). CT-undersøkelsen avdekket et stort intramuskulært hematom i musculus rectus femoris på høyre side og fettvevsreaksjon lateralt over høyre thorax og overarm tilsvarende områdene med hudblødninger. Pasienten mottok blodtransfusjon og ble etter hvert overflyttet til medisinsk intensiv for videre utredning og behandling.
Pasienten hadde klinisk blødningstendens med atraumatiske hud- og muskelblødninger, og isolert forlenget APTT. Funnene kunne være forenlig med defekt i den indre koagulasjonskaskaden og repeterte målinger bekreftet forlenget APTT i fravær av heparin. Videre ble det rekvirert faktoranalyser til undersøkelse på Oslo Universitetssykehus (OUS), Rikshospitalet som bekreftet faktormangel med en sterkt redusert faktor VIII-aktivitet på 4.4 IU/dL (50-150). På mistanke om en ervervet blødersykdom ble det utført APTT blandetest* i påvente av inhibitortiter. Testen avdekket lett forlenget APTT ved 0 timer (38s) og mer forlenget etter 1 time inkubering (54s). Funnet passet med tilstedeværelse av et inhiberende antistoff mot koagulasjonsfaktor VIII (FVIII), som etter hvert ble bekreftet ved måling av inhibitortiter på 12 BE/ml (Bethesda-enheter/ml) forenelig med ervervet hemofili A.
På bakgrunn av klinikk og laboratoriefunn ble det etter råd fra OUS, Rikshospitalet startet behandling med aktivert protrombinkomplekskonsentrat (aPCC, produktnavn FEIBA®), traneksamsyre og immunsuppresiv behandling i form av prednisolon og syklofosfamid. Pasienten responderte godt på behandlingen de påfølgende dagene, med fravær av nye blødninger og økt faktor VIII-nivå. APCC ble avsluttet etter totalt fire behandlingsdager og pasienten ble deretter flyttet tilbake til sengepost. Videre utredning avdekket sterkt positiv anti-nuclear matrix protein 2 (NXP2) og tilstedeværelse av antinuklære antistoffer (ANA). Funnet kan være assosiert med dermatomyositt (DM) eller polymyositt (PM) og er hos voksne også assosiert med underliggende kreftsykdom (1). Pasienten hadde imidlertid ingen kliniske tegn til DM eller PM og CT thorax, abdomen, bekken samt øvrige tumormarkører ga ikke holdepunkter for underliggende malignitet. Det ble ikke utført muskelbiopsi pga. blødningsrisiko.
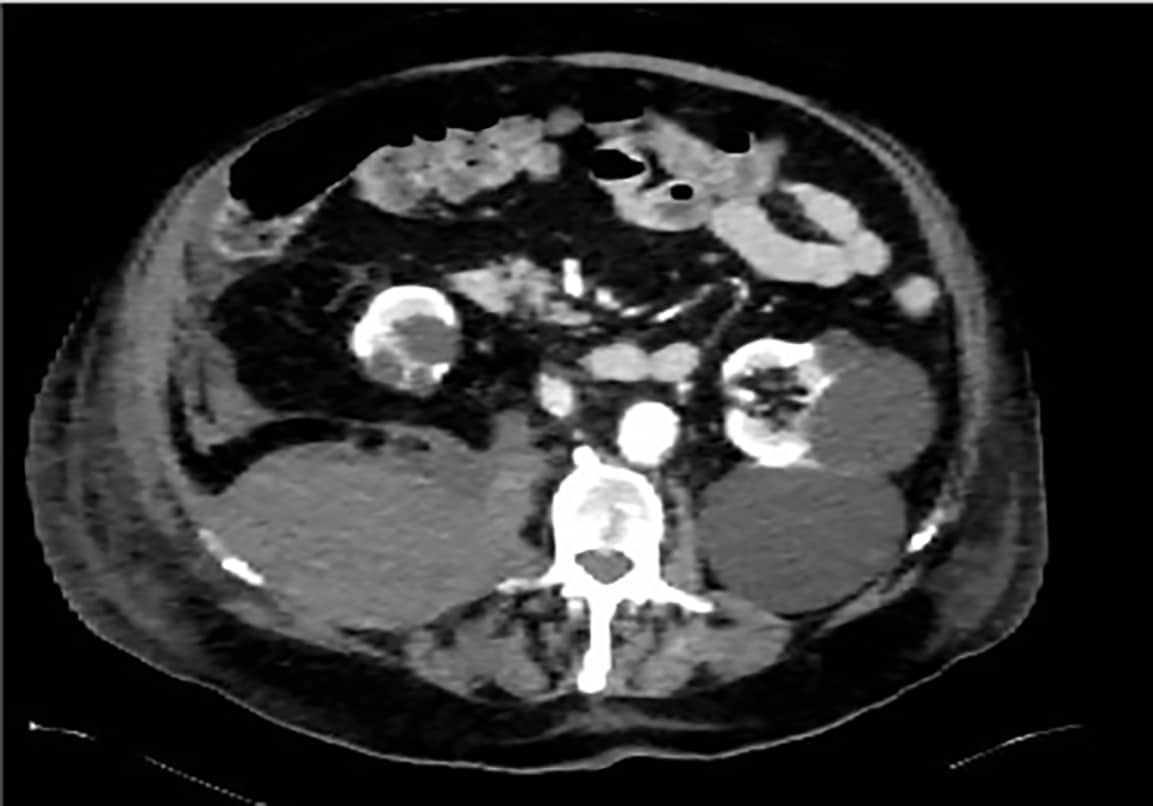
Tidlig om morgenen èn uke senere tilkalles medisinsk vakthavende til sengeposten da pasienten har utviklet akutte magesmerter. Ved tilsyn er pasienten blek, kaldsvett, smertepreget og hemoglobinnivået har falt fra 11.4-9.6 g/dl siden kvelden før. Det utføres en rask CT abdomen som avdekker et nytilkommet stort retroperitonealt hematom, sannsynlig utgående fra høyre musculus psoas, med ekstravasering på flere steder. Pasienten dekompenserer raskt og overføres tilbake til medisinsk intensiv for behandling med blodtransfusjoner, traneksamsyre og aPCC. CT-bildene konfereres med ulike spesialister, men avdekker ingen blødende kar tilgjengelig for intervensjon. Pasientens tilstand forverres ytterligere gjennom formiddagen, og det blir vurdert at pasienten ikke vil tåle et eventuelt operativt inngrep. Med hensyn til pasientens alder, prognose og begrensede behandlingsalternativer, avsluttes all pågående behandling til fordel for lindrende behandling i livets sluttfase. Pasienten sovner etter hvert inn senere samme natt, totalt to uker etter innkomst.
Ervervet hemofili er en alvorlig blødersykdom forårsaket av autoantistoffer mot koagulasjonsfaktorer, i de aller fleste tilfeller mot FVIII, og tilstanden kalles da ervervet hemofili A (AHA). Det er en svært sjelden tilstand med en insidens på 1-2 tilfeller per million (2) og rammer som regel eldre med en median alder på 74 år (3). Sykdommen er i de fleste tilfeller idiopatisk, men om lag halvparten av tilfellene er assosiert med en underliggende tilstand (3). De hyppigst assosierte tilstandene omfatter autoimmun sykdom og underliggende malignitet (3), men det ser også ut til at ervervet hemofili kan opptre ved infeksjoner, hudlidelser, under svangerskap og ved bruk av enkelte medikamenter (4). De vanligste symptomene omfatter spontane hud- og muskelblødninger samt kraftige blødninger etter venepunksjon og kirurgiske inngrep. Gastrointestinale og urogenitale blødninger er også vanlig, men i motsetning til medfødt hemofili ses leddblødninger sjeldent (3-4). Omkring 85% av pasientene får livstruende blødninger (5), og sykdommen har en mortalitet på 20% hos pasienter >65 år (6).
Klinisk blødningstendens med isolert forlenget APTT bør gi mistanke om medfødt eller ervervet faktormangel. Diagnosen ervervet hemofili stilles ved påvisning av faktormangel og tilstedeværelsen av en inhibitor mot den samme koagulasjonsfaktoren (6). I påvente av inhibitortiter kan det gjøres en APTT blandetest for å sannsynliggjøre tilstedeværelsen av antistoffer. Nivået av autoantistoffer måles i Bethesda-enheter (BE) der èn Bethesda-enhet (1 BE) tilsvarer mengden antistoffer som reduserer målt faktor VIII-nivå med 50% når den blandes med normalplasma (7).
AHA behandles med tilskudd av koagulasjonsfaktorer for å stanse blødning og immundempende behandling for å eradikere autoantistoffer. Hos en nydiagnostisert pasient med lave inhibitortiter (<5 BE) og mindre alvorlige blødninger, kan substitusjonsbehandling med FVIII-konsentrat i høye doser (100-200 E/kg) forsøkes initialt for å overkomme inhibitoren (6). I tilfeller med større blødninger eller inhibitortiter >5 BE er ikke faktorkonsentrat virksomt nok. Her må man bruke såkalte «bypassing agents» som aPCC og rekombinant faktor VIIa (rFVIIa, produktnavn NovoSeven®). APCC virker ved å tilføre små mengder aktivert faktor II, VII, IX og X, mens rFVIIa inneholder rekombinant faktor VIIa (6). De to er sidestilte førstevalg ved AHA (4,6). Behandlingen bør suppleres med traneksamsyre for å sikre god hemostase (6). Ved manglende blødningskontroll på bypassing agents anbefales i nordiske retningslinjer tilleggsbehandling med porcine plasma FVIII (pFVIII, produktnavn Obizur®), denne er imidlertid ikke tilgjengelig i Norge (6).
Eradikasjonsbehandlingen omfatter immundempende behandling med prednisolon i monoterapi eller i kombinasjon med enten syklofosfamid eller rituksimab. Førstelinjebehandlingen vil typisk være prednisolon 1-2 mg/kg gitt over 6 uker før rask nedtrapping kombinert med syklofosfamid 1.5-2 mg/kg over 3-4 måneder med samtidig profylakse mot pneumocystis jirovecii. De nordiske retningslinjene anbefaler prednisolon kombinert med enten syklofosfamid eller Rituksimab 375 mg/m2 ukentlig i 4 uker. (6), men behandlingstradisjon og klinisk erfaring tilsier å avvente med anti-CD20 behandling til andre linje. Dette støttes også av data fra det europeiske registeret over ervervet hemofili, hvor 80% av pasientene oppnår komplett remisjon ved behandling med kortikosteroider og syklofosfamid, uten økt effekt ved tillegg av Rituximab (8) Ved manglende respons i løpet av tre uker (vedvarende blødninger eller manglende økning i FVIII) kan syklofosfamid eller rituksimab (den som ikke ble benyttet i første linje) legges til, alternativt kan azatioprin eller ciklosporin forsøkes (6).
25% av pasientene med ervervet hemofili oppnår spontan remisjon mens 48% oppnår langvarig remisjon på kortikosteroider i monoterapi og 70% langvarig remisjon på steroider i kombinasjon med syklofosfamid (8). Komplett remisjon defineres som normalt nivå av faktor VIII uten målbar inhibitor (4)
Differensialdiagnosene til ervervet hemofili omfatter primært andre ervervede blødersykdommer. Disse kan deles inn i sykdommer som affiserer koagulasjonssystemet (f.eks. leversykdom og disseminert intravaskulær koagulasjon), sykdommer som affiserer blodplatene (f.eks. ervervet von Willebrands sykdom og ITP) og sykdommer som affiserer blodkar (f.eks. vaskulitter og vitamin C mangel). Isolert forlenget APTT kan også ses ved lupus antikoagulant, men denne tilstanden gir ikke klinisk blødningstendens.
Ervervet hemofili er en alvorlig sykdom hvor det er viktig med rask behandling. Da sykdommen som oftest rammer multimorbide eldre, kan tilstanden være lett å overse. Hos eldre pasienter med klinisk blødningstendens er det derfor viktig å rekvirere både APTT, INR, fibrinogen og trombocytter. Blødningstendens kombinert med isolert forlenget APTT uten åpenbar forklaring bør gi mistanke om ervervet hemofili og utløse videre utredning med faktoranalyse og inhibitortiter. Utredning og behandling av ervervet hemofili krever spesialkompetanse og bør gjøres i samråd med kompetansesenter for hemofili.
Pasientens pårørende er informert om at denne artikkelen publiseres og at de har også gitt tillatelse til bruk av bildene.



