
Anne Naalsund.
Av Anne Naalsund, Overlege (pensj), Lungeavdelingen, Oslo Universitetssykehus (OUS), Rikshospitalet. Trond Mogens Aaløkken, Overlege, Radiologisk avdeling OUS, Rikshospitalet. Tone Sjåheim, Overlege, Lungeavdelingen, OUS, Rikshospitalet
Gruppen diffuse parenkymatøse lungesykdommer (DPLS) omfatter mer enn 200 tilstander, de fleste har lik klinisk debut, men de har et vidt spekter av årsaker, forløp og prognose. Utvikling av nye behandlingsprinsipper gjør det nødvendig med nøyaktig utredning av pasienter med mistenkt DPLS. Et nært samarbeid mellom lungemedisiner, radiolog og patolog er påkrevet for å stille korrekt diagnose.
Diffus parenkymatøs lungesykdom (DPLS) er en heterogen gruppe sykdommer karakterisert ved inflammasjon og/ eller fibrose i lungeparenkymet. Gruppen omfatter mer enn 200 ulike tilstander som rubriseres sammen fordi de har visse kliniske, radiologiske og patologiske likheter (1, 2). Samtidig er de ulike, siden celler som dominerer inflammasjonen er forskjellige, og fordi stadier og utbredelse av fibrose varierer. Rask diagnostikk og behandling av DPLS kan forebygge progresjon til irreversibel lungefibrose og utvikling av respirasjonssvikt.
Tradisjonelt har begrepet ”diffus interstitiell” vært anvendt på disse sykdomsgruppene. Det parenkymatøse lungeinterstitiet er vevet som grenser mot epitelet i alveolene og endotelet i karsengen, og som inneholder lungens bindevevselementer og noen få andre celler. DILS affiserer nesten aldri bare interstitiet. Tilstandene starter oftest i epitelet i alveolene (alveolitter), i endotelet i kapillærene (vaskulitter) eller i små luftveier (bronkiolitter), undertiden også i pleura. Noen svært sjeldne sykdommer er karakterisert ved eksudat i alveolene men inkluderes også, for eksempel alveolær proteinose eller akutt interstitiell pneumoni. ”Diffus interstitiell lungesykdom” gir derfor et uriktig inntrykk av anatomisk presisjon, og internasjonalt anvendes ofte gruppebetegnelsen ”Diffus parenkymatøs lungesykdom (DPLS)”, som også er valgt i det følgende.
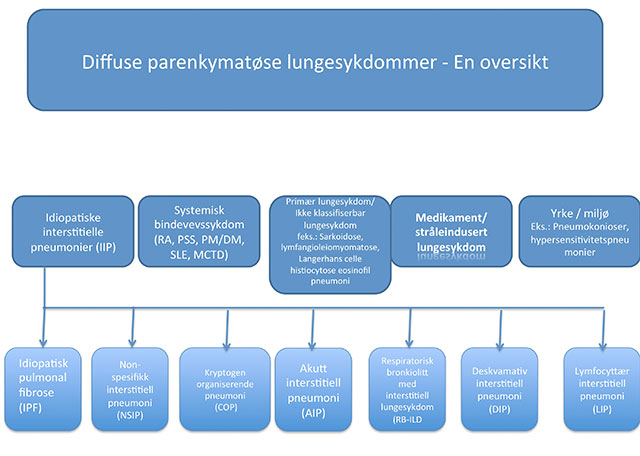
Figur 1: Klinisk klassifikasjon av diffuse parenkymatøse lungesykdommer.
Figur 1 viser en oversikt over kliniske grupper av DPLS. Av disse forekommer sarkoidose og idiopatisk pulmonal fibrose (IPF) hyppigst, mens de mest sjeldne som alveolær proteinose eller alveolær mikrolithiasis har en insidens < 1 per 1million (4-7).
IPF utgjør 40 til 60 % av idiopatiske interstitielle pneumonier (IIP)(8, 9). Gruppen IIP har de siste årene vært gjenstand for stor interesse i medisinsk litteratur, og ble derfor omtalt særskilt i nummer 2 av ”Indremedisineren” i år. Det er nylig (september 2013) publisert nye internasjonale retningslinjer vedrørende klassifisering av disse sykdommene (10).
DPLS kan være assosiert med de fleste systemiske bindevessykdommer som for eksempel reumatoid arthritt eller systemisk lupus erythematosus (SLE) og kan være første manifestasjon på disse tilstandene.
DPLS av kjent årsak omfatter også medikamentindusert lungesykdom/ stråleskader samt inhalasjon av ulike agens (inorganisk /organisk støv) ved eksponering i yrke og/eller miljø.
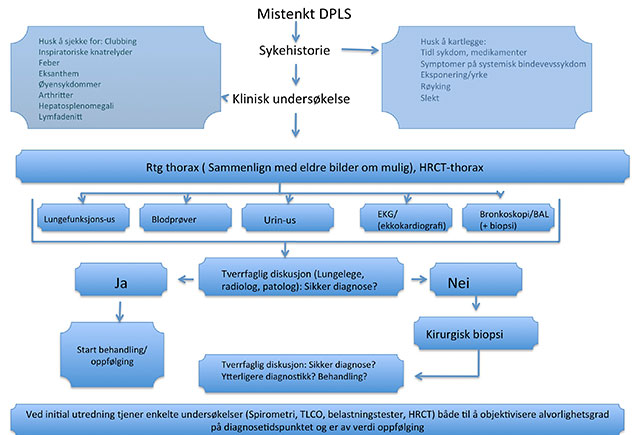
Figur 2: En skjematisk oversikt over diagnostisk tilnærming til mistenkt DPLS. Utredningen skal både bidra til å stille sikker diagnose, og til å evaluere objektivt sykdommens alvorlighetsgrad. Dessuten må komplikasjoner som pulmonal hypertensjon utelukkes. Modifisert etter Dempsey OJ, Kerr KM,
Remmen H: How to investigate a patient with
suspected interstitial lung disease. BMJ 2010 June 9;340:c2853
Utgangspunktet er oftest pasienten som oppsøker primærlegen pga åndenød, og der klinisk undersøkelse avdekker inspiratoriske knatrelyder over lungene (11). Eventuelt kan et røntgenbilde vise utbredte bilaterale fortetninger som tilfeldig funn.
Før en omfattende utredning planlegges og organiseres, må viktige differensialdiagnoser som hjertesvikt, infeksjon eller malignitet elimineres. Ved fortsatt mistanke om DPLS er en nøyaktig sykehistorie retningsgivende. Figur 2 gir en skjematisk oversikt over utredningsstrategi ved DPLS. Betydningen av at kliniker, patolog og radiolog arbeider tett sammen, framgår av skjemaet.
Nesten alle pasienter med DPLS har debutsymptomer i form av funksjonsdyspne og oftest tørrhoste.
Varighet av symptomene kan gi viktig informasjon. Symptomer i mindre enn 4 uker kan tale for akutt interstitiell pneumoni (AIP), akutt hypersensitivitetspneumoni (HP) eller kryptogen organiserende pneumoni (COP). Ved disse tilstandene vil en tidligere frisk pasient kunne utvikle invalidiserende dyspne, oftest med respirasjonssvikt i løpet av få dager.
Langt oftere utvikles åndenød ved DPLS gradvis over lang tid – måneder til år – som ved IPF og sarkoidose. Ved sarkoidose kan symptomene øke og avta over tid mens de oftest progredierer ved IPF.
Allmennsymptomer kan være uttalte, som økt trettbarhet, slapphet og vekttap ved sarkoidose, vaskulitter og ved de fleste varianter av IIP.
Mange systemiske bindevevssykdommer har ekstrapulmonale symptomer som arthritter/ artralgier, myalgier, hudforandringer, nevrologiske fenomener eller øyesymptomer. Gastroøsofageal refluks kan forverre hoste ved DPLS – det diskuteres sogar om refluks er en årsaksfaktor ved utvikling av IPF (12).
Systemiske bindevevssykdommer er assosiert med utvikling av DPLS. Likeledes sees affeksjon av andre organer som hud, nyre, hjerte ved vaskulitter (granulomatose med polyangiitt (GPA)(tidl. Mb Wegener), eosinofil granulomatose med polyangiitt (EGPA) (tidl. Churg Strauss) og kjempecellevaskulitter). Kjent astma og allergisk rhinitt kan tale for utvikling av eosinofil pneumoni, mens nasal polypose og astma er assosiert med EGPA.
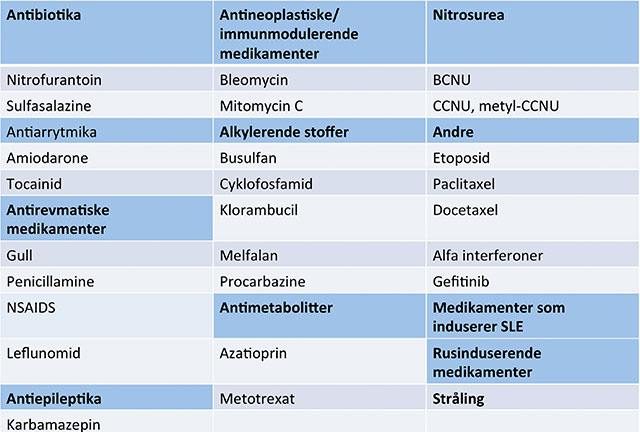
Figur 3: Oversikt over noen medikamenter som er assosiert med utvikling av diffus parenkymatøs lungesykdom.
Mange medikamenter kan forårsake utvikling av DPLS (Figur 3 ). Dette gjelder også naturmedisiner og medikamenter fra det illegale marked. Listen over medikamentrelaterte lungesykdommer øker stadig, og stilt overfor en pasient med bilaterale lungefortetninger der en slik sammenheng ikke kan utelukkes, bør det gjøres litteratursøk for nærmere informasjon. Nettstedet www.pneumotox.com kan da være nyttig. Her finnes oversikter både over medikamenter som har pulmonale bivirkninger og over histologiske mønstre utløst av ulike medikamenter. Visse preparater, for eksempel metotrexat, er assosiert med ulike histologiske forandringer i lungeparenkymet, fra en relativt benign hypersensitivitetspneumoni til utvikling av diffus alveolær lungeskade og alvorlig repirasjonssvikt. Forutsatt at sammenhengen erkjennes tidlig, er likevel de fleste former for medikamentinduserte lungesykdommer reversible.
Selv om de fleste DPLS er sporadiske, har enkelte en underliggende genetisk basis. Økt forekomst hos spesielle raser og i nær familie er velkjent ved sarkoidose. Ved enkelte sjeldne medfødte syndromer sees utvikling av lungefibrose (13).
Hos ca 50 % av pasienter med lymfangioleiomyomatose (LAM) er sykdommen assosiert med den autosomal dominante tilstanden tuberøs sklerose (14). Denne diagnosen kan stilles ved genetisk testing
Eksponering for ulike agens gjennom yrket, i nærmiljøet eller ved reisevirksomhet er forbundet med utvikling av DPLS. Inhalasjon av uorganisk støv som asbest, kull, silica og beryllium kan føre til parenkymatøs lungesykdom, ofte mange år etter at eksponeringen fant sted. Eksponering for organisk støv som termofile sopper (treskerlunger, kornhandlerlunger, luftfukterlunger), visse bakterier (atypiske mykobakterier) eller dyreproteiner (fugleholderlunger) på arbeidsplassen eller i hjemmemiljøet er en viktig årsak til hypersensitivitetspneumonier.
Sigarettrøyk er assosiert med utvikling av IPF, og også med respiratorisk bronkiolitt assosiert med interstitiell lungesykdom (RB-ILD) og deskvamativ interstitiell pneumoni (DIP). Pulmonal Langerhanscelle histiocytose – en sjelden lungesykdom karakterisert ved utvikling av granulomer og cyster i lungeparenkymet – sees bare hos røykere (15).
Aktiv røyking kan bidra til komplikasjoner ved Goodpastures syndrom. Det er antatt at røyking ødelegger alveolveggen, og at dette bidrar til at den alveolære basalmembranen blir tilgjengelig for sirkulerende antibasalmembran antistoffer (16).
Cyanose ved DPLS er sjelden på diagnosetidspunktet fordi dette reflekterer alvorlige forstyrrelser i gassutvekslingen, med O2-metning < 90 %.
Pasienter med akutt interstitiell pneumoni (AIP), akutt hypersensitivitetspneumoni eller alvorlig grad av kryptogen organiserende pneumoni har imidlertid ofte utviklet leppecyanose/ neglesengcyanose allerede ved første legekontakt. Fingerclubbing, eller hypertrofisk osteoarthropati, sees særlig ved IPF (50 – 60 %), men også ved DIP. Ved sarkoidose er derimot clubbing svært sjelden. Påvisning av hudblødninger kan tale for vaskulitt, mens ulike eksanthemer kan sees ved progressiv systemisk sklerose (PSS), polymyositt/ dermatomyositt (DM/PM), SLE eller ved sarkoidose.
Ved auskultasjon over lungene er inspiratoriske knatrelyder et dominerende funn ved IPF og ved støvlunger som asbestose. Pasienter med sarkoidose har derimot upåfallende lungefysikalia selv ved avansert sykdom. Inspiratoriske knatrelyder og samtidig pipelyder i ekspiriet kan høres hos pasienter med hypersensitivitetspneumoni men også ved bronkiolitt.
Kardial undersøkelse
Ved undersøkelse av hjertet vil en aksentuert 2. hjertetone over pulmonalostiet gi mistanke om utvikling av pulmonal hypertensjon, enten som komplikasjon til DPLS eller pga primær karaffeksjon som ved PSS eller ved PM/DM. Ved enkelte systemsykdommer kan kardial status avdekke arrytmier eller holdepunkter for kardiomyopati (sarkoidose, EGPA, RA)
Leddaffeksjon tydende på artritt taler helst for en systemisk bindevevssykdom, oftest RA, SLE eller MCTD mens artralgier uten objektive leddfunn er hyppigere ved sarkoidose.
Ofte vil en god sykehistorie og resultater av klinisk undersøkelse gi pekepinn om aktuell diagnose. Videre utredning (figur 2) skal verifisere diagnosen og objektivisere funksjonsnivået på diagnosetidspunktet. En slik objektiv ”basalstatus” er nyttig når behandlingseffekt og sykdomsforløp skal vurderes.

Figur 4: Aktuelle blodprøver/ urinprøver som bør taes ved mistanke om DPLS.
Blodprøver kan være av diagnostisk verdi i utredning av DPLS (figur 4), men en negativ test kan aldri utelukke en spesifikk diagnose.
Eosinofili kan tale for eosinofil granulomatose og polyangiitt (EGPA), eller for eosinofil pneumoni. Lymfopeni sees ved aktiv sarkoidose og hyperkalsemi hos 5-15 % av disse.
Serum angiotensinkonverterende enzym (s-ACE) har lav sensitivitet i diagnosen av sarkoidose, og britiske retningslinjer angir at ACE målinger har begrenset plass i diagnostikk av lungesarkoidose (2).
Antineutrofilt cytoplasmatisk antistoff (ANCA) er økt i et cytoplasmatisk mønster hos pasienter med granulomatose med polyangiitt (GPA) (tidl mb Wegener), og i et perinukleært mønster hos pasienter med EGPA.
Revmatoid faktor og en autoimmun profil bør foretaes hos pasienter med DPLS av ukjent årsak fordi lungeaffeksjon kan være første manifestasjon på disse systemsykdommene.
Påvisning av presipiterende antistoffer av type IgG, særlig mot muggsopp og fugleantigener, kan – på tross av lav sensitivitet og spesifisitet–være av verdi i diagnostikk av hypersensitivitetspneumonier.
HIV-testing kan være nyttig fordi enkelte former for DPLS, som lymfocytær interstitiell pneumoni kan være assosiert med immunsvikt. Lungeinfiltrater ved pneumocystis jiroveccii pneumoni som sees hos immunkompromitterte pasienter, kan mistolkes som DPLS. (Fig.5)
Inflammatoriske markører (CRP, SR) vil hos enkelte være forhøyet på diagnosetidspunktet, og kan derfor være nyttige senere i forløpet for å vurdere behandlingsrespons.
Hematuri eller proteinuri kan tale for en tilgrunnliggende vaskulitt eller systemsykdom.
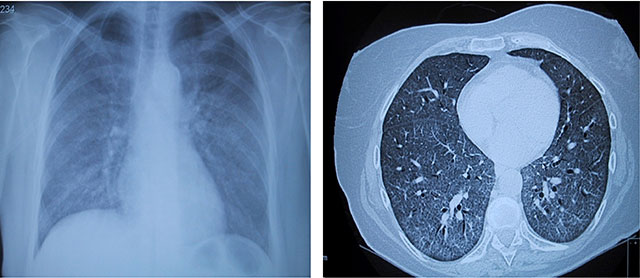
Figur 5: Betydning av HIV-test i diagnostikk av pasienter med diffus parenkymatøs lungesykdom. 65 år gammel kvinne, tidligere frisk. I løpet av måneder utviklet økende dyspne, etterhvert respirasjonssvikt. Innlagt til utredning av bilaterale lungefortetninger. Røntgen thorax viser lungefortetninger, diffust sløret preg. HRCT: Generelt økt tetthet i lungeparenkymet med mattglasspreg. Videre utredning viste pneumocystis jirovecci infeksjon. HIV test var positiv.
Rtg thorax kan være nyttig initialt i utredningen fordi ca 90 % av pasientene med DPLS har et patologisk bilde på diagnosetidspunktet. Et normalt bilde utelukker dermed ikke DPLS (Fig. 6)
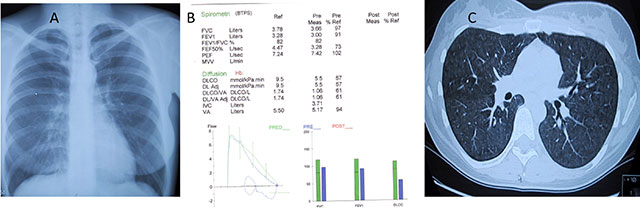
Figur 6: 31 år gml kvinne utredet for funksjonsdyspne av noen måneders varighet.
A. Normalt røntgen thorax.
B. Lungefunksjonsundersøkelser viste normal belgfunksjon bedømt ved spirometri, men redusert transferfaktor for CO (TLCO).
C. HRCT var patologisk med utbredte centrilobulære noduli med mattglasspreg i begge lunger best forenlig med subakutt
hypersensitivitetspneumoni. Hadde positive presipiterende antistoffer for fugleantigener (IgG).
Basale fortetninger med fibrose og skrumpning kan tale for IPF, men dersom det i tillegg sees pleurale fortetninger, eventuelt med forkalkede plaques er asbestose en mer sannsynlig diagnose.
Høy-oppløsnings-CT thorax (High Resolutin CT, HRCT) som benytter 1-2 mm tynne snitt gir bedret oppløsning sammenlignet med konvensjonell CT, og dermed bedret karakterisering av utbredelse, lokalisasjon og mønster av forandringene i parenkymet. Indikasjon for HRCT thorax i utredning av DPLS vil være (16,17):
– Påvise DPLS hos symptomatiske pasienter der rtg thorax er beskrevet som normalt, eventuelt med usikre funn.
– Begrense aktuelle differensial-diagnostiske muligheter.
– Påvise egnet område for utførelse av kirurgisk lungebiopsi.
– Evaluere behandlingsrespons.
Undertiden er HRCT bildet så karakteristisk for en gitt tilstand at diagnosen kan stilles ut fra dette alene og kirurgisk lungebiopsi utelates (figur 7, figur 8). Bilateralt forstørrede lymfeknuter i hilus eller mediastinum, men uten affeksjon av fremre mediastinum er nær patognomonisk for sarkoidose.

Figur 7: Forutsatt relevant klinikk: Noen tilstander der HRCT kan være diagnostisk.
Bronkoskopi med aspirasjon av bronkoalveolærskyllevæske (BAL)er ved siden av HRCT thorax et viktig hjelpemiddel i diagnostikk av DPLS. Mens bronkialaspirat består av materiale fra sentrale luftveier og er nyttig i diagnostikk av cancer og infeksjoner, reflekterer funn i korrekt utført BAL forholdene i alveolene. Det er nylig i regi av The American Thoracic Society (ATS) utarbeidet detaljerte retningslinjer for bruk av BAL i diagnostikk av DPLS (18). Undersøkelsen gjennomføres i lokalbedøvelse i forbindelse med bronkoskopi. Analyser av BAL kan i visse tilfeller være diagnostisk, eventuelt redusere differensialdiagnostiske alternativer. Lymfocyttdominans i væsken kan tale for hypersensitivitetspneumoni, sarkoidose eller kryptogen organiserende pneumoni (COP) og analyse av CD4/CD8 T-celleratio er av verdi for å skille hypersensitivitetspneumonier og sarkoidose. Eosinofil dominans sees ved eosinofil pneumoni, eosinofil bronkitt eller medikamentindusert lungesykdom. Ved alveolær proteinose har BAL-væsken ofte et melkeaktig utseende, og den er positiv ved farging med periodisk-acid-Schiff (PAS-farging).
Ved tilstander der diffuse alveolære blødninger dominerer, som ved småkarvaskulitt eller ved idiopatisk pulmonal hemosiderose, vil cytologisk undersøkelse vise hemosiderinfylte makrofager også om sykdommen er inaktiv på undersøkelsestidspunktet; eventuelt vil væsken være blodig ved pågående blødning.
Funn av > 5 % Langerhansceller (CD-1a positive) i BAL gir sterk mistanke om pulmonal Langerhanscelle histiocytose (PLHCH), men kvantitative kriterier for diagnosen er ikke endelig fastsatt. Ved elektronmikroskopi vil funn av såkalte Birbeck granula i Langerhansceller være diagnostisk.
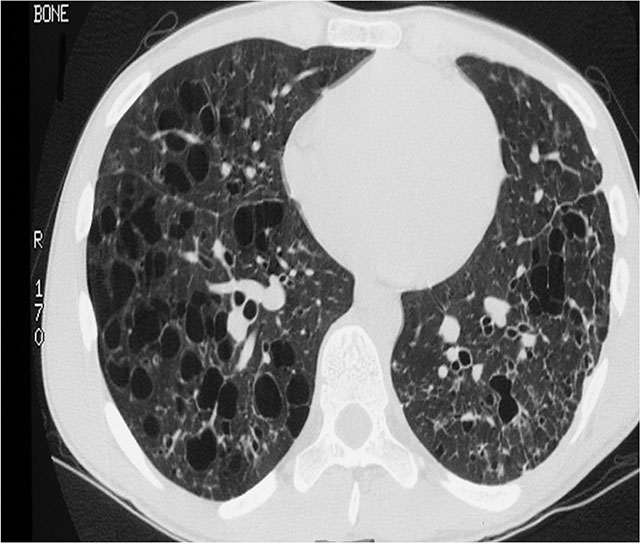
Figur 8: Pulmonal Langerhanscelle histiocytose (PLHCH). 16 år gammel gutt, røykt i 2 år, siste halvår repeterende episoder med pneumothorax. HRCT viste multiple irregulære cyster med varierende veggtykkelse, mest uttalt i øvre lungeavsnitt samt mikronoduli forenlig med LHCHC. PLHCH er en lungesykdom av ukjent årsak karakterisert ved granulomer med Langerhansceller som infiltrerer og destruerer terminale bronkioler og fører til hulrom-
dannelse og cyster i lungeparenkymet-
Tverrfaglig vurdering i team med radiolog, patolog og lungelege har ført til at færre pasienter henvises for kirurgisk lungebiopsi. Likevel vil det være behov for biopsitaking i de situasjoner der kombinasjonen av serologi, BAL og HRCT thorax ikke har ført til diagnostisk avklaring. Transbronkiale lungebiopsier (TBB) og endobronkiale biopsier som utføres under bronkoskopi, er nyttige i diagnostikk av malignitet og granulomatøse lungesykdommer som sarkoidose. TBB kan også være diagnostisk ved akutt eosinofil pneumoni, kryptogen organiserende pneumoni og ved subakutt hypersensitivitetspneumoni. Undersøkelsen har derimot ingen plass i diagnostikk av IPF. Endobronkial/ endoskopisk ultralydveiledet biopsi av mediastinale lymfeknuter (EBUS / EUS) er nyttig ved mistanke om sarkoidose, og for å utelukke malignitet (19). Hvorvidt det velges å gjøre transbronkial biopsitaking eller EBUS/EUS i diagnostikk av sarkoidose avhenger av fasiliteter ved de enkelte sykehus – diagnostisk presisjon er uansett ca 90 %.
Ved de øvrige parenkymatøse lungesykdommer er en større vevsbit påkrevet for sikker diagnostikk, og inngrepet gjøres i slike tilfeller oftest ved videoassistert thorakoskopi (VATS), eventuelt ved minithorakotomi. Fordi patologiske forandringer ofte er heterogene og multifokale, kreves biopsier fra flere steder, helst fra forskjellige lungelapper. Dette bedrer både sensitivitet og spesifisitet. Biopsistedet velges på bakgrunn av funn på HRCT, og bør være fra lungevev utenfor men nær områder med åpenbar parenkymdestruksjon/ fibrose.
Selv om lungenes mekaniske egenskaper og gassveksling påvirkes ved DPLS, er lungefunksjonsmålinger av liten diagnostisk verdi. Derimot er slike målinger av stor betydning når det gjelder å bedømme sykdommens alvorlighetsgrad på diagnosetidspunktet og også ved seinere kontroller for å bedømme prognose og behandlingsrespons.
Det karakteristiske mønstret ved utvikling av DPLS er restriksjon med lav forsert vitalkapasitet (FVC) og redusert forsert ekspiratorisk volum på 1 sekund (FEV1) i samme forhold som FVC . Under oppfølging vil et fall i FVC > 10 % i løpet av 6 måneder være signifikant hos en pasient med IPF, og assosiert med en betydelig økt mortalitetsrisiko.
For å få et inntrykk av gassveksling under hvile og arbeid benyttes måling av transfer faktor for karbonmonoksyd (TLCO), arteriell blodgass/oksymetri, 6-minutters gangtest og eventuelt sykkelergometri. Disse undersøkelsene gir informasjon om pasientens funksjonsnivå, og ved kontroller, om sykdommens utvikling.
TLCO gir et non-invasivt estimat for gassvekslingen i lungene og avtar i prinsippet proporsjonalt med reduksjon i funksjonelle alveolokapillære enheter (20). De laveste verdiene – ofte < 20 % av forventet verdi – sees hos pasienter med kombinert IPF/emfysem, ved cystisk lungesykdom (LAM, PLHCH) eller ved utvikling av pulmonal hypertensjon. Betydelig forhøyede verdier sees hos pasienter med vaskulitt og pågående alveolære blødninger fordi CO da taes opp i alveolært hemoglobin.
Måling av arteriell blodgass i hvile vil hos de fleste pasienter med DPLS vise normale funn langt ut i forløpet, og først i avansert stadium hypoksemi og hypokapni.
Ved arbeidsbelastning sees derimot ofte fall i O2-metningen, selv ved beskjeden grad av fysisk aktivitet. Ved 6-minutters gangtest måles oksygenmetning før-, under og etter aktivitet, i tillegg til total ganglengde. Enkelte pasienter med DPLS kan virke klinisk upåfallende i hvile med normal spirometri og upåfallende blodgasser, men utvikler alvorlig grad av hypoksemi i forbindelse med gangtest. Anstrengelsesutløst hypoksemi er korrelert til utvikling av pulmonal hypertensjon som kan komplisere DPLS (21). Slike pasienter bør henvises til ekkokardiografi, eventuelt til høyre hjertekateterisering som er gullstandard for måling av pulmonalarterie trykket.
Sykkelergometri har størst verdi når det er tvil om diagnosen, og gjennomført test viser seg å være normal. Pasienten har da ikke DPLS.
I mange europeiske land tas disse pasientene hånd om ved spesialklinikker med tverrfaglig kompetanse (lungelege, radiolog, patolog, thoraxkirurg, revmatolog), flere steder også med tilgang på helsepersonell utdannet i palliativ omsorg. Et slikt teamsamarbeid er antatt å bedre diagnostikk, oppfølging, kanskje også prognose for disse pasientene (9, 22,23, ).
I Norge er det enighet om en ”teamtilnærming” til disse pasientene, men foreløpig primært på regionsnivå. Lokale lungeavdelinger og primærhelsetjenesten er likevel sentrale i samarbeidet for å bedre oppfølgingen av pasienter med DPLS.
Pasienter der utredning for lungetransplantasjon kan være aktuelt, bør henvises OUS, Rikshospitalet tidlig i sykdomsforløpet – iallfall ved signifikant fall i FVC (> 10 % fra utgangsverdi i løpet av de første 6 – 12 måneder), ved fall i TLCO i samme tidsrom > 15 %, og ved utvikling av pulmonal hypertensjon. Internasjonale retningslinjer anbefaler dessuten at pasienter med IPF inkluderes i multisenterstudier der dette er mulig (22).
Det har derfor i løpet av det siste året pågått et nordisk samarbeid for å opprette databaser primært for pasienter med IPF, men på sikt også for andre pasienter med IIP, både for å sikre bedre epidemiologiske data enn hva vi har i dag, men også for å ha pasienter tilgjengelig for inkludering i aktuelle studier.



