
Eystein S. Husebye.
Av Eystein S. Husebye, Overlege professor dr med, Klinisk institutt 2, Universitetet i Bergen og Endokrinologisk seksjon, Medisinsk avdeling, Haukeland Universitetssykehus.
Immunsystemet skal beskytte oss mot invasjon av fremmede organismer, men samtidig tolerere seg selv. Sentralt er den unike plastisiteten i dannelsen av B- og T- celle reseptorer med millioner av ulike spesifisiteter (1). I utviklingen av immunsystemet foregår det en intrikat seleksjonsprosess der immunceller som reagerer på eget vev selekteres bort, mens de med reaktivitet for fremmede vev, organismer og substanser beholdes. Dette skille er ikke absolutt og kontrollmekanismer er etablert for å hindre aktivering av de autoreaktive cellene som finnes. De siste 20 årene har brakt mye ny kunnskap om rollen til sentral immunologisk toleranse ved autoimmune sykdommer. Helt sentralt i denne forskningen har studier av den sjeldne, monogent, arvelige sykdommen autoimmunt polyendokrint syndrom type 1 (APS-1) vært.
APS-1, også kalt autoimmun polyendokrinopati – kandidiasis – ektodermal dystrofi (APCED) er karakterisert av organ-spesifikk autoimmunitet mot en rekke organer (Fig. 1). Hovedmanifestasjonene er hypoparathyreoidisme, primær binyrebarksvikt (Addisons sykdom) og kronisk mukokutan kandidiasis; 2 av disse 3 hovedkomponentene kreves tradisjonelt for diagnose (2). Andre endokrine organer som ofte også rammes, er ovarier med menopause i tenår eller tidlig voksen alder, og de insulinproduserende betacellene med type 1 diabetes som resultat. Autoimmun thyreoideasykdom, som er svært vanlig i befolkningen ellers, ses sjelden. Men organaffeksjon er ikke bare begrenset til endokrine kjertler: Gastrointestinale manifestasjoner som autoimmun enteropati med malabsorpsjon, autoimmun gastritt med vitamin B12- og jernmangel, og autoimmune hepatitt er også vanlig (3). Likeledes har mange pasienter sykdommer fra ektodermale vev, slik som alopesi, vitiligo og emaljehypoplasi. Sistnevnte ses hos flertallet av pasientene. Keratitter er en fryktet komponent i sykdommen som kan føre til blindhet. Også de ektodermale komponentene, antar man er autoimmunt betinget, selv om autoantigener så langt bare er funnet i samband med alopesi og vitiligo.
Lenge framstod det som et paradoks at pasientene hadde kroniske kandidainfeksjoner på hud og slimhinner, altså et tegn på immunsvikt. Nyere undersøkelser har vist at pasientene har autoantistoffer mot komponenter i immunforsvaret mot kandida, spesielt mot interleukin (IL) 17F og IL-22 (4). En autoimmunt betinget «kortslutning» i forsvarskjeden mot kandidainfeksjoner kan derfor forklare hvorfor pasientene er utsatt for dette.
I 1997 ble sykdomsgenet funnet og publisert samtidig av 2 konkurrerende forskningsgrupper (5, 6), og kalt autoimmun regulator (AIRE), et navn som har vist seg å være dekkende for genproduktets funksjon. AIRE uttrykkes kun i noen få spesialiserte celler i kroppen, først og fremst i medullære epiteliale celler i tymus der AIRE regulerer gentranskripsjon. AIRE er ikke en tradisjonell transkripsjonsfaktor som regulerer uttrykket til et begrenset antall gener, men har en overordnet regulatorisk rolle, der AIRE skrur på tusenvis av gener, spesielt de som koder for vevsspesifikke proteiner. På den måten eksponeres umodne T-celler for tusenvis av egne proteiner som ellers bare finnes i diskrete organer. Autoreaktive T-celler som reagerer på eget vev («selv») kan da elimineres eller omdannes til regulatoriske T celler. Ved APS-1 er denne sorteringsmekanismen defekt. Autoimmune T-celler unnslipper til sirkulasjonen og kan siden gi opphav til autoimmun sykdom (7, 8). Hvorfor binyrebarken og paratyreoideakjertlene er spesielt utsatt for dette angrepet er imidlertid ikke kjent.
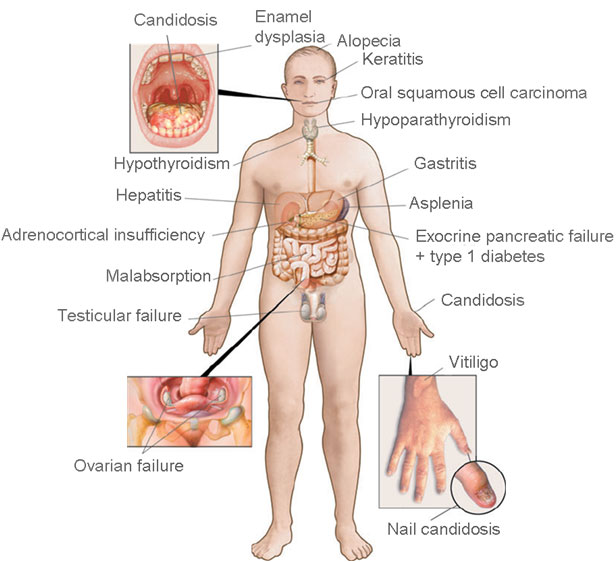
Figur 1. Homunculus med kliniske manifestasjoner hos pasienter med APS-1.Figuren er gjengitt med tillatelse fra Husebye et al. Journal of Internal Medicine, 265, 514-529, 2009.
Det klassiske forløpet er debut av kronisk kandidiasis i tidlig barnealder (0-4 år), etterfulgt av hypoparatyreoidisme (4-8 år) og binyrebarksvikt (8-12 år), men mange kan debutere med andre manifestasjoner, og det kan gå mange år mellom komponentenes debut (3, 9). Derfor diagnostiseres mange sent, og noen aldri. Vi har eksempler på at mange ikke har fått diagnosen før i voksen alder, og i mange familier er det rapporter om barn som har dødd i barnealder der årsaken kan ha vært akutt binyrebarksvikt eller hypokalsemiske kriser (9). I snitt utvikler pasientene 4 ulike komponenter, men det er stor fenotypisk variasjon, også innenfor samme familie. I Norge finnes APS-1 med en frekvens på 1:80 000, mens den er betydelig vanligere i enkelte befolkningsgrupper som blant finner (1:25 000), sardinere (1:14000) og persiske jøder (1:9000). I hver av de sistnevnte befolkningene er det en bestemt mutasjon i AIRE som dominerer. I dag kjenner vi til identiteten til om lag 50 pasienter i Norge, av dem er 35 i live. Vanlige dødsårsaker er akutt binyrebarksvikt og malignitet. Karsinomer i munnhule og spiserør er overrepresentert ved APS-1, og er assosiert til kronisk ubehandlet kandidiasis, spesielt i kombinasjon med røyking.

I 2006 oppdaget Tony Meager og medarbeidere at APS-1 pasienter hadde svært høye nivåer av antistoffer mot type 1 interferoner, noe som tidligere bare var beskrevet hos pasienter med tymom og sent debuterende myastenia gravis (10). Måling av autoantistoffer mot interferon omega har vist seg å være en svært sensitiv og spesifikk test på APS-1 som nå benyttes som en diagnostisk prøve. Alle de kjente norske pasientene har slike autoantistoffer; positivitet er rapportert allerede før 1 års alder (11) og autoantistoffnivåene holder seg konstant i flere 10-år. I tillegg har APS-1 pasientene autoantistoffer mot en rekke andre vevsspesifikke proteiner som er korrelert til organmanifestasjoner i organene de er uttrykt, for eksempel «side-chain cleavage enzyme», aromatisk L-aminosyre dekarboksylase (AADC), glutaminsyre dekarboksylase (GAD), tryptophan hydroksylase type I/II (TPH I/II) og «NACHT leucine rich repeat 5» (NALP5) (Tabell 2). I Norge tilbys analyse av disse autoantistoffene ved Hormonlaboratoriet på Haukeland Universitetssykehus.

Etter at AIRE ble klonet, ble det straks satt i gang undersøkelser om det var genforandringer i AIRE også ved vanligere autoimmune sykdommer som type 1 diabetes og primær binyrebarksvikt. Man fant ikke noen klar sammenheng, men man testet da bare på de vanligste mutasjonene og gjorde ellers «single nucleotide polymorphism» (SNP)-analyser. Senere har det vist seg at kombinasjonen thyreoiditt og systemisk sklerose er assosiert med genvarianten V301M (12); vi publiserte tidligere at en pasient med binyrebarksvikt hadde denne varianten (13). Området der denne variasjonen er lokalisert, er essensiell for funksjonen til AIRE og regulerer bindingen til blant annet histoner. V301M-varianten har en frekvens på 1 promille i flere populasjoner. Nylig publiserte data exomdata fra om lag 60 000 individer fra Broad Institute i Cambridge (Exome aggregation consortium, http://exac.broadinstitute.org/) viser at det finnes en rekke variasjoner i AIRE med frekvenser omkring 1 promille. Til sammen kan kanskje slike variasjoner disponere for vanlige former for autoimmunitet med familiær opphopning, men det gjenstår å vise.
Studier av monogene sykdommer har vist seg å være et ekstremt nyttig verktøy for å forstå fysiologi og sykdomsmekanismer. Kartlegging av AIRE sin funksjon har lært oss mye om mekanismen for sentral immunologisk toleranseutvikling og hvordan feil i denne kan lede til autoimmun sykdom. APS-1 er sjelden, men underdiagnostisert. Måling av autoantistoffer mot interferon omega er en god screeningprøve. Diagnosen stilles enten ved typisk klinikk (2 av 3 hovedmanifestasjoner) eller påvisning av sykdomsfremkallende AIRE-mutasjoner.



