
Tone Sjåheim
Av Tone Sjåheim, overlege dr.med. May Brit Lund, overlege dr.med. Avdeling for spesialisert lungemedisin, OUS Rikshospitalet
Idiopatisk lungefibrose er en progressiv fibroserende lungesykdom av ukjent årsak som primært rammer eldre (>60 år). Sykdommen har dårlig prognose og det er ingen kurativ behandling. Nye medikamenter kan bremse sykdomsprogresjonen, og det er derfor viktig at diagnosen stilles tidlig i forløpet. HR-CT thorax kan være diagnostisk, evt. kreves åpen lungebiopsi.
Interstitielle lungesykdommer er tidligere omtalt i Indremedisineren [1, 2]. Idiopatisk lungefibrose (IPF) er den vanligste av de idiopatiske interstitielle pneumoniene og kjennetegnes av rask sykdomsprogresjon, alvorlig prognose og manglende respons på behandling med immundempende medikamenter. Den inflammatoriske komponenten er ikke dominerende og fibrosen dannes uten vesentlig forutgående inflammasjon. Det er viktig å skille IPF fra andre typer interstitiell lungesykdom med fibrose for å kunne forutsi prognose og valg av behandling. Den tidligere betegnelsen kryptogen fibroserende alveolitt skal ikke lenger brukes.
Ifølge internasjonale retningslinjer er IPF definert som en spesifikk form av kronisk, progressiv fibroserende interstitiell pneumoni av ukjent årsak som forekommer hyppigst hos voksne mellom 50 og 70 år, og er assosiert med det histopatologiske og/eller radiologiske bilde av UIP (usual interstitial pneumonia) [3].
Det foreligger ikke pålitelige estimater for insidens og prevalens av IPF i Norge. Utenlandske undersøkelser angir årlig insidens 7-16/100.000. Insidensen øker med alder og IPF er vanligst i 60 -70 års alder. Median alder ved diagnosetidspunkt er ca 66 år. Tilstanden ses hyppigst hos menn og de fleste er tidligere røykere. IPF forekommer sjelden hos personer under 50 år. Familiær IPF forekommer.
IPF kan ha akutt eller subakutt debut. Symptomene kommer vanligvis gradvis, over få måneder til år. Ved tidlige stadier kan kliniske funn være fraværende, men bilaterale fine inspiratoriske knatrelyder over basale deler av lungene er ofte et tidlig funn [4]. IPF bør mistenkes hos eldre med progredierende funksjonsdyspne og/eller vedvarende tørrhoste som ikke kan forklares av annen årsak, som f.eks kols eller hjertesvikt. Hos enkelte er intraktabel hoste dominerende. Allmennsymptomer som generell sykdomsfølelse, tretthet og vekttap kan forekomme, mens feber, ledd- og muskelsmerter er uvanlig og tyder på annen tilstand. I sent stadium utvikles ofte pulmonal hypertensjon.
Ved mistanke om IPF bør pasienten så tidlig som mulig henvises til spesialist i lungesykdommer for videre utredning.
Anamnese: Grundig anamnese er viktig for å utelukke andre årsaker som kan gi tilsvarende kliniske, radiologiske og histopatologiske funn, og bør omfatte familieanamnese, yrkes- og miljøeksponering, røykevaner, nåværende og tidligere medikamentbruk og om det foreligger bindevevssykdom/autoimmun sykdom. Ved mistanke om medikamentrelatert lungesykdom er det nyttig å sjekke nettstedet www.pneumotox.com.
Lungefunksjon: Objektiv kartlegging med spirometri, måling av gassdiffusjon (DLCO), statiske lungevolum, seks minutter gangtest og arteriell blodgass er nødvendig. Forsert vitalkapasitet (FVC) er den vanligste parameteren som følges ved IPF. Det er stor variasjon i sykdomsforløpet [5], men de fleste pasienter opplever gradvis forverring med årlig fall i FVC rundt 0,1 – 0,2 liter. En mindre andel kan ha betydelig raskere fall i lungefunksjon, mens noen kan være stabile i flere år.
Radiologi: HR-CT thorax har en sentral plass i utredning av interstitiell lungesykdom og kan være diagnostisk ved påvisning av UIP-mønster. Undersøkelsen må være av tilfredsstillende teknisk kvalitet med 1 mm snitt og bedømmes av en erfaren thoraxradiolog.
Bronkoskopi: IPF diagnose kan ikke stilles ved bronkoskopi, men undersøkelsen kan være nyttig for å utelukke infeksjon og begrense differensialdiagnostiske muligheter.
Transbronkiale lungebiopsier: Erfaringsmessig gir slike biopsier for lite vev til å stille diagnosen IPF.
Kirurgisk (åpen) lungebiopsi: Dette er påkrevet for å stille diagnosen IPF dersom det på HR-CT thorax ikke påvises UIP-mønster. Det forutsetter imidlertid at pasientens lungefunksjon ikke er så redusert at et kirurgisk inngrep innebærer uakseptabel risiko.
Blodprøver: Blodprøver har ingen sentral plass i utredningen, men autoantistoffscreening (anti-CCP, ANA, ANCA og myosittspesifikke antistoffer) er viktig for å utelukke tilgrunnliggende bindevevssykdom/vaskulitt.
Diagnosen IPF krever eksklusjon av andre årsaker til interstitiell lungesykdom. Kliniske symptomer, radiologiske og histologiske funn er ikke spesifikke for IPF og tilstanden kan derfor være vanskelig å skille fra andre interstitielle lungesykdommer. Flere tilstander som kronisk hypersensitivitetspneumoni og interstitiell lungesykdom relatert til asbest, medikamenter eller bindevevssykdom kan ved langkommen sykdom også fremvise UIP-mønster. Radiologiske og evt. histologiske funn må tolkes i en klinisk sammenheng og diagnosen IPF bør stilles på sykehus hvor det holdes tverrfaglige møter med thoraxradiolog, patolog og lungelege.
Ut fra gjeldende retningslinjer [3] kan IPF diagnosen stilles dersom det påvises UIP-mønster på HR-CT thorax og andre årsaker til radiologisk UIP-mønster kan utelukkes. Radiologisk UIP-mønster er detaljert beskrevet i retningslinjene og omfatter subpleural basal dominans, retikulære forandringer, bikakemønster (honeycombing) med eller uten traksjonsbronkieektasier samt fravær av forandringer som er uforenlig med UIP-mønster (øvre dominans, uttalt mattglass, mikronoduli, konsolidering og mosaikkperfusjon /airtrapping) (figur 1). Avgjørende for UIP-mønster er påvisning av bikakemønster. Påvisning av bikakemønster er det eneste kriteriet som skiller et definitivt UIP-mønster fra et mulig UIP-mønster (figur 2). Dersom bikakemønster ikke kan påvises på CT thorax, kreves åpen lungebiopsi for å stille IPF diagnosen. Aktuelle retninglinjer [3] gir dermed mulighet til å stille IPF diagnose uten kirurgisk lungebiopsi først når det foreligger bikakemønster – som er et uttrykk for langt fremskredet sykdom med irreversibel fibrose. Dagens retningslinjer som krever bikakemønster for diagnose, er av den grunn kritisert og under revisjon.
Indikasjon for åpen lungebiopsi via thorakotomi eller videoassistert thorakoskopi må vurderes nøye i hvert enkelt tilfelle og nytteverdi veies opp mot risiko. Et kirurgisk inngrep i narkose hos IPF pasienter kan utløse akutte forverringer og et ARDS lignende bilde. Kirurgisk biopsi anbefales derfor ikke ved sterkt redusert lungefunksjon (DLCO < 40 % av forventet verdi) eller hos pasienter over 65 år. Mange pasienter med sannsynlig IPF, og mulig UIP-mønster på CT thorax, får derfor ingen endelig diagnose og fratas derved mulighet for medikamentell behandling.
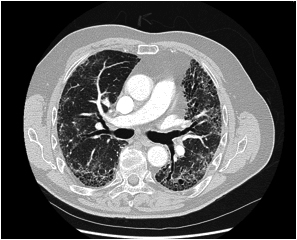
Figur 1: HR- CT thorax med definitivt UIP-mønster hos 72 år gammel mann med gradvis økende funksjonsdyspne og hoste. Bildet viser retikulære forandringer, bikakemønster (honeycombing) og traksjonsbronkieektasier og det er fravær av forandringer som er uforenlig med UIP-mønster HR-CT thorax er dette tilfelle diagnostisk og åpen lungebiopsi er ikke nødvendig. Diagnose: IPF, da øvrige årsaker til UIP-mønster kunne utelukkes.
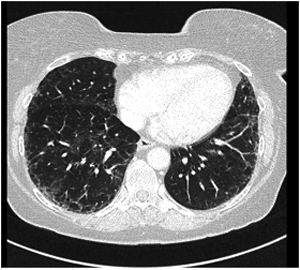
Figur 2. HR- CT thorax med mulig UIP-mønster hos 60 år gammel kvinne med funksjonsdyspne og hoste. Bildet viser grovt retikulært mønster med enkelte små cyster og perifere bronkieektasier, men ikke bikakemønster. Flekkvise mattglassfortetninger. Det er ikke funn som utelukker UIP. Radiologisk mest sannsynlig diagnose er non-spesifikk interstitiell pneumoni (NSIP). Kirurgisk lungebiopsi viste histologisk UIP-mønster. Diagnose: tverrfaglig diskusjon konkluderte med IPF, da øvrige årsaker til UIP-mønster kunne utelukkes.
De siste tiår har det vært betydelig forskning vedrørende medikamentell behandling av IPF, men de fleste studier har kommet negativt ut. Prednisolon har ingen plass i behandlingen, verken alene eller i kombinasjon med andre immundempende medikamenter. Eneste unntak er ved akutte eksaserbasjoner hvor det er vanlig å behandle med høydose kortikosteroider. Imidlertid foreligger det ikke data fra kontrollerte studier. Kombinasjonsbehandling med steroider, azathioprin og acetylcystein var tidligere anbefalt standardbehandling. Denne behandlingen ble imidlertid frarådet i 2012 etter at det ble påvist økt hospitalisering og mortalitet hos pasienter med slik trippelbehandlingen sammenlignet med monoterapi med acetylcystein eller placebo [6]. I påvente av mer dokumentasjon har monoterapi med acetylcystein (antioksidant) vært i bruk inntil nylig, men det er nå vist at atacetylcystein ikke har effekt på fall i lungefunksjon [7].
To ulike medikamenter er nå godkjent for behandling av IPF, Esbriet (pirfenidon) og Ofev (nintedanib). Esbriet er klassifisert som et immunsuppressivt middel med antifibrotiske og antiinflammatoriske egenskaper og har vært i bruk i Norge siden februar 2012. Ofev er et antineoplastisk middel (tyrosinkinasehemmer) som nylig ble registrert i Norge (april 2015). Begge medikamentene har vist at fallet i FVC ble redusert med vel 100 ml etter 1 år sammenlignet med placebogruppene [8-10]. Esbriet eller Ofev anbefales til IPF pasienter med mild til moderat sykdom (def. FVC > 50 % og DLCO > 30 % av forventet verdi). Det foreligger ikke studier på pasienter med dårligere lungefunksjon. Det er ingen øvre grense for oppstart av behandling. Behandlingsmål er å stabilisere sykdommen og forsinke progresjon. Det er derfor viktig at diagnosen IPF stilles tidlig i forløpet, før det oppstår irreversible fibrose. Pasientene må imidlertid informeres om at behandlingen ikke forventes å bedre eller kurere sykdommen.
Behandlingen bør startes og følges opp av spesialist i lungesykdommer som har erfaring med diagnostisering og behandling av IPF. Begge medikamentene er kostbare (NOK 340.000/år) og det må søkes om individuell refusjon. Pasientene må følges tett, spesielt ved oppstart av behandlingen, med jevnlig kontroll av lungefunksjon og registrering av bivirkninger. Det pågår en rekke medikamentstudier og det forventes flere behandlingsmuligheter for IPF pasienter.
Da IPF kan ha rask sykdomsprogresjon bør pasienter < 65 år henvises Rikshospitalet allerede ved diagnosetidspunktet for vurdering av mulighet for lungetransplantasjon.
Ved tidlige stadier av sykdommen er lungefunksjonen ofte normal. Etter hvert utvikles en restriktiv ventilasjonsinnskrenkning og redusert gassdiffusjon. IPF pasienter har lenge normal arteriell blodgass i hvile, men desaturerer markert ved fysisk anstrengelse. Først ved langt fremskreden sykdom utvikles kronisk respirasjonssvikt. Median overlevelse ved IPF angis til 2-3 år etter at diagnosen er stillet. Omtrent 10 % av IPF pasienter rammes av akutt forverring. IPF eksaserbasjon har dårlig prognose med > 90 % mortalitet innen 6 måneder. Ugunstige prognostiske faktorer for overlevelse er DLCO < 40 % av forventet verdi ved diagnosetidspunktet, uttalt fibrosedannelse på HR-CT thorax, desaturering til SaO2 < 88 % ved 6 min gangtest og pulmonal hypertensjon [11].
Interessekonflikter:
Tone Sjåheim: mottatt foredrags-honorar fra InterMune og Boehringer Ingelheim Norway KS. May Brit Lund: ingen.
Referanser



