I desember 2021 publiserte vi sammen med kolleger fra flere medisinske fagfelt en oppdatert veileder for amyloidose på nettsidene til ulike fagmedisinske foreninger og www.Helsebiblioteket.no (1). Målet med en slik veileder er å rette søkelys mot tilstanden slik at amyloidose mistenkes og utredning igangsettes der det er relevant. Det er viktig å avgjøre hvilken type amyloidose som foreligger og om det dreier seg om systemisk eller lokalisert affeksjon. Vi gjennomgår her når det er viktig å tenke på amyloidose i mage-tarmkanalen og lever, og hvordan man kan påvise og eventuelt behandle denne.
Svein Oskar Frigstad, Medisinsk avdeling, Vestre Viken Bærum Sykehus
Melinda Raki, Avdeling for patologi, Oslo universitetssykehus
Tale Norbye Wien, Medisinsk avdeling, Vestre Viken Bærum Sykehus
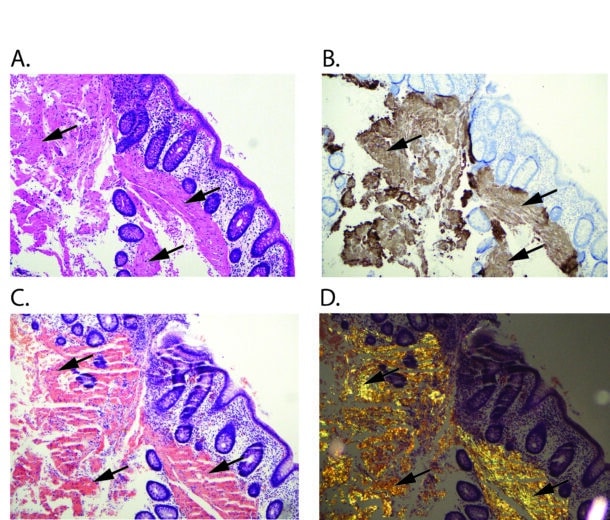
Amyloidose er en gruppe sykdommer som kjennetegnes ved avleiring av polypeptidmateriale ekstracellulært i organer og vev. Dette materialet kalles amyloid, og betegnelsen stammer fra gresk amylon, som betyr stivelse, og kommer av at avleiringene har egenskaper som minner om stivelse i histologiske snitt. Hovedkomponenten i amyloid er amyloide fibriller, som er endestadiet i en sekvensiell forandring i polypeptidets folding som resulterer i en form som er karakteristisk for amyloidose. Det amyloide fibrilleproteinet varierer med typen av amyloidose, men de ulike fibrilleproteinene har alle en karakteristisk og felles ultrastruktur (2). Farging med kongorødt benyttes rutinemessig for å påvise amyloid i vevsprøver (figur 1).
Dannelsen og avleiringen av amyloid forårsaker sviktende organfunksjon ved å fortrenge normale vevskomponenter og vevsfunksjoner. Avleiringen er en dynamisk prosess som potensielt kan være reversibel, men gir ofte irreversibel organskade. Amyloid kan avleires i alle kroppens organer, enten i ett organ (lokalisert amyloid) eller i flere organsystemer (systemisk amyloidose). Det er stor variasjon i klinisk presentasjon, progresjon og prognose. Typiske, men likevel uspesifikke, manifestasjoner ved systemisk amyloidose er bl.a. vekttap, slapphet, tretthet, proteinuri med eller uten nyresvikt, restriktiv kardiomyopati, gastrointestinale og respiratoriske symptomer, hepatomegali, hudaffeksjon ev. med purpura, polynevropati og karpaltunnelsyndrom.
Populasjonsbaserte studier med epidemiologiske data for insidens og prevalens av amyloidose har vært sparsomme, og norske tall foreligger ikke. Tall fra svenske sykehusregistre i perioden 2001-2008 viser insidens av ervervet og arvelig systemisk amyloidose på henholdsvis 8.3 og 2.8 per million personer/år (3, 4). Ut fra data fra autopsistudier og dødsårsaksregistre er det sannsynlig at amyloidose er underdiagnostisert og at et betydelig antall pasienter ikke får adekvat diagnostikk og behandling (5). Økende rapportert insidens av systemisk amyloidose internasjonalt de siste årene antas å skyldes økende oppmerksomhet om sykdommen (6, 7).
De ulike typer av amyloidose klassifiseres ut fra fibrilleproteinet, det protein som danner de amyloide fibrillene. Amyloidtypen forkortes med stor A for amyloid, med påfølgende forkortelse for det angjeldende proteinet, f.eks. AA for amyloid dannet fra proteinet serum amyloid A (SAA), ATTR for amyloid dannet fra transtyretin (TTR), og AL for amyloid dannet fra lette immunglobulinkjeder. Ytterligere spesifisering kan gis etter proteinnavnet, f.eks. ATTRwt eller ATTRv (wt = villtype og v = variant). Videre inndeler man vanligvis amyloidosene etter deres deponeringsmønster i systemiske og lokaliserte former og ut fra arvelige og ervervede former.
Per i dag kjenner vi 36 ulike proteiner som kan danne amyloide fibriller i mennesket (8). Av disse opptrer 18 som systemisk amyloidose og 22 som lokalisert amyloidose, og for noen proteiner finnes både systemiske og lokaliserte former. Identifisering av det amyloide fibrilleproteinet, dvs. typing av amyloidosen, samt å fastslå om det er systemisk eller lokalisert sykdom er helt avgjørende for riktig behandling og kan ha stor betydning for sykelighet og overlevelse. De ovennevnte tre typene, dvs. AA, ATTR og AL amyloidose utgjør over 90% av alle tilfeller av systemisk amyloidose.
Den diagnostiske prosessen kan summeres i fire trinn: 1) mistanke om amyloidose ut fra symptomer og tegn, 2) verifisering av amyloid i vevsprøve, 3) etiologisk diagnose, dvs. typing og 4) kartlegging av organutbredelse. Påvisning skjer i dag dessverre ofte ikke før i avanserte stadier av sykdommen når mye av organfunksjon kan være tapt, og behandlingsmulighetene er begrenset. En viktig forutsetning for en tidlig diagnose er å ha kjennskap til sykdommen slik at man starter utredning når det er indisert.
Gullstandard for påvisning av amyloid er farging av vevsprøve med kongorødt. Ved sparsomme avleiringer og ved usikkert resultat av kongorød farging kan immunfarging for amyloid-P brukes for å bekrefte amyloidavleiring. Sikrest og nyttigst informasjon får man ved å direkte biopsere det vevet som er mistenkt affisert. Sikrest fordi sensitiviteten er større, og nyttigst fordi det sikkert definerer at det angjeldende organ er affisert. Hvis dette ikke lar seg gjøre, har indirekte biopsi, ofte fettvevsbiopsi fra abdomen tradisjonelt vært anbefalt (9). Tradisjonelt har man brukt immunhistokjemi for typing av amyloidproteinet. Metoden er pålitelig for påvisning av AA amyloidose og kan ofte brukes mtp ATTR amyloidose, men er kjent for å ha lav sensitivitet for AL amyloidose. Massespektrometrisk analyse er en relativt ny metode som ble først tatt i bruk for amyloidtyping ved Mayo klinikken for om lag 15 år siden. Analysen gjøres oftest på avleiringer som er skåret ut fra formalinfiksert vev med laserdisseksjon, har meget høy sensitivitet og kan også avdekke sjelden forekommende og nye amyloidproteiner (10).
Anamnese er viktig. Vi må spørre detaljert om symptomer på organaffeksjon. Familiehistorie må kartlegges med hensyn på ev. arvelige amyloidoser. Spesielt for AA-amyloidose er det viktig med tidligere inflammatoriske sykdommer og rusanamnese som kan disponere for AA-amyloidose (11). I den kliniske undersøkelsen og prøvetaking skal man undersøke for affeksjon av de organsystemer som vanligvis rammes av systemisk amyloidose, spesielt hjerte- og nyreaffeksjon, med ødemer, forhøyet pro-BNP og troponin, samt proteintap i urin. Husk også tegn på nerveaffeksjon som ortostatisk hypotensjon, karpaltunnelsyndrom, perifer nevropati, i tillegg til tarm- blære- og ereksjonsfunksjonsforstyrrelser (5, 12). Påvisning av en eventuell monoklonal komponent er sentralt med tanke på AL-amyloidose, og man bør gjøre proteinelektroforese med immunfiksasjon samt kvantitering av frie lette kjeder kappa og lambda (s-FLC). Ekkokardiografi gjøres ved mistanke om hjerteaffeksjon, og hvis AL amyloidose er utelukket går man videre med amyloidose-skjelettscintigrafi med tanke på ATTR hjerteamyloidose (1, 5).
Mange pasienter med amyloidose har sammensatte symptombilder, med affeksjon av flere organsystemer. Et godt tverrfaglig samarbeid i utredning, behandling og oppfølging er viktig.
Flere typer amyloidose kan affisere mage- og tarmkanalen, oftest AA, AL eller ATTR (13). Affeksjon av mage-tarmkanalen er rapportert i 3-8 % av pasienter med systemisk AL amyloidose (14, 15). Nesten to tredjedeler av pasienter med arvelig form av ATTR amyloidose rapporterer symptomer fra fordøyelsessystemet, mens ATTRwt sjelden gir slike symptomer (16). I tidlig fase av sykdommen er det sjelden vesentlige symptomer fra mage eller tarm, og subklinisk affeksjon kan foreligge. Mekanismene som gir symptomer er ofte sammensatt. Manifestasjoner av sykdommen er relatert til dysfunksjon og skade av det autonome nervesystem, og således kan symptomer i hele fordøyelseskanalen oppstå (17). Det er rapportert både tidlig metthetsfølelse, kvalme, oppkast og vekttap fra øvre deler av fordøyelsessystemet, og vekslende obstipasjon og diare ved tarmaffeksjon. Magesmerter vil ofte være til stede, ofte etter matinntak. Symptomene kan ofte være betydelige og er forbundet med redusert livskvalitet og dårligere prognose (17). Det er imidlertid ingen sikker korrelasjon mellom symptombyrde og alvorlighetsgrad av sykdommen i tidlig fase, og det vil ofte være vekttap og andre allmensymptomer som leder oss til diagnosen. I senere stadier av sykdommen forekommer ofte feilernæring og mangeltilstander utløst av malabsorpsjon (18). Massiv gastrointestinal blødning kan forekomme ved uttalt amyloidavleiring som gir skjøre blodkar (19).
Diagnostikk er avhengig av adekvat utredning og prøvetaking. Symptomer kan likne andre tilstander som ofte vil være utelukket. Cøliaki, inflammatorisk tarmsykdom og kroniske infeksjoner må utelukkes. Blødning fra tarmslimhinnen med påvisbart blod i avføring er sjelden i tidlig fase. Amyloidose vil heller ikke gi utslag på inflammasjonsmarkører som ofte måles i utredning av tarmsykdom, for eksempel fekal kalprotektin (20). Bildediagnostikk slik som CT abdomen eller MR av tynntarm vil ofte ikke vise signifikante funn. Ved endoskopiske undersøkelser kan slimhinnen se helt normalt ut. Det er derfor viktig å ta biopsier til spesialfarging (kongorødt) for å stille diagnosen. Dette er oftest ikke rutine ved patologiske avdelinger, og samarbeid mellom kliniker og patolog er viktig for å avklare diagnosen. Biopsier fra rektum har vært mest brukt, da de er lettest tilgjengelige, men amyloid avleiring kan påvises i alle deler av fordøyelseskanalen. Det er viktig med dype biopsier som omfatter submukosa (20).
Funksjonsundersøkelser av øsofagusmotilitet, ventrikkeltømming og tarmmotilitet er ofte patologiske, og kan avklare behovet for tiltak. Ved dysfagi, bør øsofagusmanometri utføres. Ventrikkeltømmingstest er best tilgjengelig med 13C-pusteprøve eller funksjonsultralyd, selv om ventrikkelscintigrafi fortsatt regnes som gullstandard i utredning av gastroparese (21). En trådløs motilitetskapsel (smart-pill) er også tilgjengelig ved enkelte sykehus. Røntgen transittid kan gi god informasjon om funksjon av tykktarm.
Det kliniske bildet ved amyloidose i lever er oftest relatert til symptomer fra systemisk amyloidose slik som vekttap og redusert almentilstand. Leveren er affisert hos 90 % av pasienter med AL amyloidose og 60 % av pasienter med AA amyloidose (22). Leveren kan være moderat til betydelig forstørret, og ofte er ALP betydelig forhøyet (23). Graden av hepatomegali samsvarer ikke direkte med grad av amyloidavleiring (24).
Ved ultralyd av lever kan amyloidose gi et bilde med forstørret lever med heterogen ekkogenisitet, men dette er ikke et spesifikt funn. Ved CT kan det noen ganger ses fokale endringer i attenuasjon eller forkalkninger. MR kan gi økt signalintensitet av leverparenkymet ved T1 vekting (sannsynligvis grunnet småkarsykdom) uten vesentlige endringer ved T2 vekting (25). Diagnosen stilles sikkert kun ved leverbiopsi med spesialfarging. Det er rapportert økt forekomst av blødning etter leverbiopsi ved amyloidose, men dette regnes i praksis ikke for å være et problem ved gode rutiner for observasjon.
Kronisk leversykdom med leversvikt og portal hypertensjon ved amyloidose i lever er sjelden. Ascites kan forekomme, men er oftere relatert til hjertesvikt og systemisk sykdom enn avansert leversykdom. Leveraffeksjon er imidlertid assosiert med dårligere prognose, og behandlingen retter seg hovedsakelig mot systemsykdommen (23).
Mange pasienter med amyloidose har sammensatte symptombilder, med affeksjon av flere organsystemer. Et godt tverrfaglig samarbeid i utredning, behandling og oppfølging er viktig. Behandlingen består av symptomreduserende tiltak og sykdomsspesifikk behandling.
Symptomreduserende tiltak for amyloid i mage-tarm-kanalen innebærer kostveiledning. Små og hyppige måltider anbefales. Ved gastroparese anbefales lavt innhold av fett og fiber samt forsiktighet med alkohol og niktotin (21). Flytende, kvernet og most mat tømmes lettere fra magesekken. Metoklopramid eller erytromycin kan være aktuell som symptomatisk behandling for å stimulere ventrikkeltømming. Dersom hovedproblemet er kvalme, kan antiemetika uten prokinetiske egenskaper benyttes, for eksempel ondansetron. Ved diare eller obstipasjon behandles disse på vanlig måte med tarmregulerende midler. Ved behov for næringstilførsel, kan både sondenæring og parenteral næringstilførsel vurderes. Forebygging og overvåkning for å forhindre mangeltilstander er en viktig del av behandlingen.
For noen typer amyloidose finnes det spesifikk behandling, for andre ikke. Behandling som virker ved målrettet å redusere tilgjengelig mengde av det proteinet som danner fibrillene, er godt etablert og er spesifikk for hver enkelt type amyloidose (5, 12). For eksempel vil AL-amyloidose, som skyldes en klonal plasmacellesykdom, behandles med kjemoterapi og evt. høydose kjemoterapi med autolog stamcellestøtte (HMAS), mens AA-amyloidose behandles ved å redusere inflammatorisk aktivitet, da SAA er et akuttfaseprotein.
I senere år er det også utviklet medikamenter som kan hemme syntese av fibrilleproteinet på gennivå, samt medikamenter som reduserer tilgjengelig fibrilledannende protein er å stabilisere proteinet i en struktur der det ikke danner fibriller, såkalt konformasjonsstabiliserende behandling for ATTR) (5, 26-29).
Arvelige amyloidoser kan behandles med levertransplantasjon i de tilfeller der leveren er ansvarlig for syntese av variant form av den amyloiddannende protein. Levertransplantasjon er nå nesten forlatt som behandling for arvelig ATTR etter at det har kommet andre alternative behandlingsformer for å redusere eller å stabilisere det fibrilledannende proteinet TTR, men fortsatt aktuelt for andre sjeldnere varianter for arvelig amyloidose (29).



