Bakgrunn. Akutt respirasjonssvikt er den vanligste årsaken til intensivmedisinsk behandling. Den oppstår hos 20-75/100 00/år, og skyldes oftest akutte betennelsesforandringer i lungevevet. Denne artikkelen gir en oversikt over klinikk, etiologi, patofysiologi og behandling ved akutt alvorlig respirasjonssvikt, med hovedvekt på svikt fremkalt av sekundære betennelsesforandringer.
Materiale og metode. Oversikten representerer ikke en systematisk litteraturgjennomgang, men bygger på forfatterens kliniske intensivmedisinske erfaring og lungesviktrelatert forskningsarbeid samt eget arkiv og avgrenset litteratursøk i Medline.
Resultater. Akutt respirasjonssvikt kan skyldes sykdom eller skade som påvirker lungene direkte. Alvorlige infeksjoner eller vevsskade andre steder i kroppen kan også forårsake slik svikt ved at aktiverte blodceller og proinflammatoriske stoffer følger blodstrømmen til lungene hvor de utløser en sekundær betennelsesreaksjon. Den sistnevnte type respirasjonssvikt er mest alvorlig, og har en dødelighet i sykehus som angis i området 30-50%.
Fortolkning. Det finnes ingen spesifikk behandling av den sekundære betennelsesreaksjonen, den er oftest reversibel når de utløsende skade- eller sykdomsprosesser går tilbake. Respiratorbehandling kan hindre at alvorlig hypoksemi gir tilleggsskader av både primært og sekundært affisert vev. Med moderne intensivbehandling skyldes bare 10-15% av dødsfallene ved akutt respirasjonssvikt selve lungesvikten. De fleste dør fordi pasientene i tillegg utvikler svikt av andre organer (multiorgansvikt).

Helge Opdahl
Av Helge Opdahl, Nasjonalt kompetansesenter for NBC* medisin, (*forkortelse for “Nuclear, Biological and Chemical”), Akuttmedisinsk avdeling, Medisinsk Divisjon, Oslo Universitetssykehus, Ullevål
Alvorlig respirasjonssvikt hos tidligere lungefriske skyldes oftest en utbredt betennelsesprosess i lungevevet. Hvis årsaken er infeksiøs (for eksempel bakteriell pneumoni) går prosessen oftest tilbake ved effektiv behandling av det utløsende agens. Ved ikke-infeksiøse årsaker, som mekanisk skade av lungevev, aspirasjon av mageinnhold til luftveiene eller inhalasjon av skadelige gasser, finnes ingen spesifikk behandling.
En betennelsesprosess i lungevev kan også oppstå sekundært til brudd- eller vevsskader andre steder i kroppen, eller til infeksjoner hvor fokus er lokalisert fjernt fra lungene. Slike prosesser er en vanlig årsak til at pasienter trenger respiratorbehandling. Tilbakegang av svikten er betinget av hvorvidt den utløsende årsak er reversibel. I denne artikkelen er det først og fremst respirasjonssvikt som oppstår sekundært til sykdomsprosesser utenfor thorax som omtales.
Grunnlaget for artikkelen er et ikke-systematisk litteratursøk i PubMed med et skjønnsmessig utvalg av artikler basert på forfatterens erfaring innen feltet, samt på artikler hentet fra forfatterens eget litteraturarkiv. Forfatteren har over 20 års erfaring fra kirurgiske intensivavdelinger, Oslo Universitetssykehus, Ullevål, og har doktorgrad fra Universitetet i Oslo i 1993 med vekt på nøytrofile granulocytters betydning for utvikling av akutt lungesviktsyndrom.
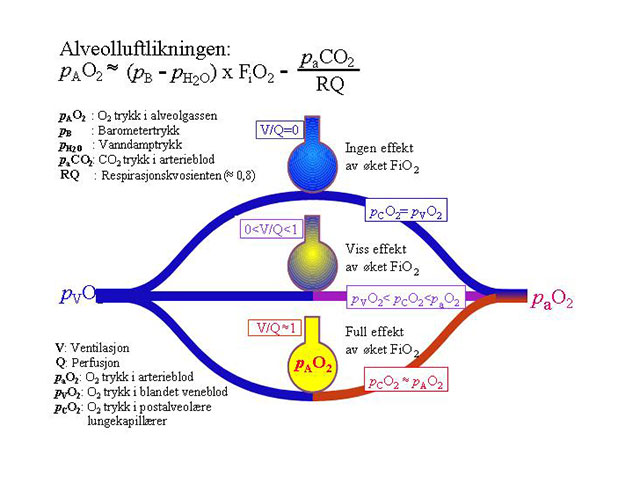
Figur 1: Alveolluft og lungeshunt
Alveolluftens oksygentrykk, pAO2, bestemmes av barometertrykk, trykket av CO2 i alveolene og oksygenfraksjonen i inspirasjonsluften. Normalt øker pO2 i forbipasserende lungekapillærer i takt med pAO2. Ved lungeshunt avhenger effekten av øket O2 i inspirasjonsluften av forholdet mellom normalt ventilerte (V/Q ≈ 1), dårlig ventilerte (O<V/Q<1) og ikke-ventilerte (V/Q=0) alveoler, dvs shuntfraksjonen i lungene. Behandling (først og fremst øket luftveistrykk) som konverterer dårlig ventilerte alveoler til godt ventilerte og ikke-ventilerte til ventilerte, eller som øker O2 innhold i blandet venøst blod (CVO2) ved å bedre forholdet mellom O2 tilbud og -forbruk, vil også øke paO2.
Lungeshunt oppstår når deler av blodet som strømmer gjennom lungene passerer lungevev hvor oksygenopptaket fra alveolene er redusert eller opphørt. Dette fører til at arterieblodets oksygentrykk (paO2) blir lavere enn det som forventes ut fra oksygentrykket i inspirasjonsluften. Forskjellen mellom aktuell og forventet paO2. avhenger av forholdet mellom de fraksjoner av blodet som passerer alveolområder med henholdsvis normal, nedsatt eller ingen ventilasjon (Fig 1). Vanlige årsaker til nedsatt ventilasjon er øket luftveismotstand eller at lungevevets elastisitet er redusert. Opphørt ventilasjon ses først og fremst i områder hvor alveolene er fylt av ødemvæske eller puss, eller hvor de har falt sammen (atelektaser).
I klinisk sammenheng angis shunten ofte som forholdet mellom oksygentrykket i arterieblod og fraksjonen av O2 i inspirasjonsluften, paO2/FiO2, eller som differansen mellom beregnet oksygentrykk i alveolene (pAO2), og det som måles i arterielt blod: pAO2–paO2.
Akutt respirasjonssvikt etter store skader og sjokktilstander, uavhengig av om skaden innbefattet lungene, har lenge vært kjent blant militærleger og skadekirurger. Tilstanden ble ofte kalt ”sjokklunge”. Den ble erkjent som et eget sykdomsbilde først etter at Asbaugh og medarbeidere beskrev den som et syndrom i 1967 (1) og kalte det ”acute respiratory distress syndome” (ARDS). På norsk er betegnelsen akutt lungesviktsyndrom. Betegnelsen økte forståelsen for at slik respirasjonssvikt ikke var en hyperakutt pneumoni, men at svikten kunne oppstå sekundært til en rekke andre sykdommer og skader som fremkaller betennelse i lungevevet.
Lungenes elastisitet er redusert, og nedsatt oksygeninnhold i blodet gir øket trettbarhet i respirasjonsmuskulaturen. Symptombildet likner pneumoni, med anstrengt ventilasjon, takypné, takykardi og cyanose. Funnene ved auskultasjon er ofte uspesifikke. En internasjonal konsensuskonferanse i 1990-årene (2) vedtok følgende definisjon av akutt lungesviktsyndrom:
– Bilaterale, diffuse lungefortetninger
– Utelukking av kardial årsak til respirasjonssvikten
– paO2/FiO2 ≤ 26,7 kPa (200 mm Hg)
Det samme sykdomsbildet, men med en mindre alvorlig lungesvikt (paO2/FiO2 ≤ 40,0 kPa (300 mm Hg)) har vært klassifisert som acute lung injury (ALI, på norsk betegnet som akutt lungeskade). ALI betegnelsen er foreslått fjernet og erstattet av ”ARDS med lavere alvorlighetsgrad”. Skadede lunger blir lett infiserte. Bakterielle pneumonier kan være utgangspunkt for sepsis, som igjen kan utløse akutt lungesviktsyndrom. Liknende respirasjonssvikt kan utløses av bl.a. rusmidler og forgiftninger (7) samt transfusjoner (8). Definisjonsmessig kan derfor både pneumoni og de fleste andre årsaker til direkte skade av lungene inngå i begrepet akutt lungesviktsyndrom (se ramme nedenfor).
I et skandinavisk materiale fra 1999 beregnet man en insidens på 17,9 tilfeller av akutt lungeskade per 100 000 personer per år (3). Data fra USA indikerer en forekomst på 78,9 tilfeller (4). Tall som angir forekomsten av akutt lungesviktsyndrom varierer med en faktor på ti mellom forskjellige land (5). Divergensene kan skyldes variasjoner i tolking av sykdomsdefinisjon, tilgjengelighet av helsetjenester, forskjeller i bruk av intensivbehandling eller i rapporterings- og registreringsrutiner. Sannsynlig forekomst er innen området 20-75 pasienter per 100 000 personer pr. år (6).
Vev som utsettes for mekanisk eller kjemisk skade, infeksjon eller hypoksi frigjør molekyler med proinflammatoriske egenskaper (9). Samtidig kan endotelets overflate endres slik at dette både induserer koagulasjon (10) og aktiverer nøytrofile granulocytter og andre leukocytter. Kombinasjonen av proinflammatoriske agens varierer med de utløsende faktorer. Bakterietoksiner, celle- eller fettfragmenter, aktiverte komplementfaktorer og pro-inflammatoriske cytokiner er viktige. I tillegg induseres endringer i celleoverflate og syntesemønster hos f. eks. endotelceller, nøytrofile granulocytter, monocytter og makrofager (11). Enhver sykdom eller skade som kan indusere en generell inflammatorisk respons, kan også utløse et akutt lungesviktsyndrom (se tekstramme) .

I akuttfasen dannes økte mengder proteinrik ødemvæske i alveolene. Dette skyldes øket permeabilitet for proteiner og væske i lungekarene. Karenes hydrostatiske trykk kan være normal. Ødemdannelsen er sjelden så massiv at ødemvæske kommer opp i de øvre luftveier. Alveolepitelets evne til å resorbere ødemvæske fra alveolene er nedsatt. Proteiner som inngår i koagulasjonskaskaden lekker ut med ødemvæsken, og gjør at denne blir som en gel når aktiverte makrofager og monocytter stimulerer koagulasjonen (11). Kombinasjonen av ødelagt cellemateriale og koagulerte plasmaproteiner danner typiske hyaline membraner i de små luftveiene (12).
Betennelsesreaksjonen gjør at nøytrofile granulocytter binder seg til endoteloverflaten og deretter vandrer ut i vevet. Når de stimuleres av proinflammatoriske agens fra blod eller fra lokale celler i lungene utskiller de store mengder reaktive oksygenforbindelser og vevsnedbrytende enzymer (13). Denne reaksjonen skader både endotel og type 1 pneumocytter i alveolene og bryter ned lungevevet. Også andre immunkompetente celler deltar i betennelsesutviklingen.
Påvirkning av bakterietoksiner og proinflammatoriske cytokiner kan endre endotelcellenes overflate fra å ha antikoagulante til å ha pro-koagulante egenskaper (10). Aktivering av trombocytter og koagulasjonskaskaden fører til trombosering i lungenes mikrosirkulasjon. Dette endrer perfusjonsforholdene og øker lungekarmotstanden med fare for høyre ventrikkelsvikt.
Etter den akutte betennelses- og ødemfasen følger en fase med resorbsjon av væske og reparasjon av ødelagt vev. Selv om noe av lungevevet forblir skadet og omdannes til arrvev, vil mesteparten av det affiserte lungevevet gjenvinne normal funksjon (14). Hos noen pasienter skjer en kraftig innvekst av bindevevsceller. Lungene hos slike pasienter forblir fibrotiske og stive selv om oksygeneringsevnen bedres. Et slikt forløp er assosiert med redusert overlevelse (14).
Pasienter som fyller kriteriene for akutt lungesviktsyndrom behandles på overvåkings- eller intensivavdelinger. Det finnes i dag ingen spesifikk profylakse mot, eller kausal behandling av, syndromet når dette skyldes en ikke-infeksiøs betennelsesprosess. Ikke-stereoide antiinflammatoriske legemidler (NSAIDs) har ingen beskyttende effekt, og påvirker ikke sykdomsforløpet (15). Kortikosteroider, som kan ha gunstig effekt hos enkeltpersoner (se nedenfor), bedrer ikke overlevelsen i større pasientpopulasjoner (16). Behandlingen bygger derfor på to hovedprinsipper:
Økning av oksygenmengden i inspirasjonsluften har ofte dårlig effekt ved akutt lungesviktsyndrom. Ikke-invasiv respirasjonsstøtte via ansiktsmaske har også begrenset effekt. Intervensjoner som øker antallet vel ventilerte alveolområder gir derimot en betydelig bedring (fig 1), og overtrykksventilasjon med respirator er hjørnestenen i behandling av alvorlig respirasjonssvikt. I akuttfasen forutsetter behandlingen vanligvis anestesi, intubasjon og dyp sedasjon. Kombinasjonen av anestesi/sedasjon og overtrykksventilasjon har ofte negative sirkulatoriske effekter hos normo- og hypovolemiske personer. Overtrykksventilasjon øker intra-torakalt trykk og nedsetter hjertets diastoliske fylling og minuttvolum, og medikamentelt betinget vasodilatasjon aksentuerer endringene. Ved uendret O2-forbruk vil redusert hjerte-minuttvolum redusere O2-innholdet i arterieblodet hos pasienter med konstant lungeshunt (Fig 1).
Moderne respiratorer har mange ventilasjonsmodi, de grunnleggende prinsippene for respiratorbehandling i akuttfasen er likevel enkle. Lungene ventileres vanligvis med tidevolum i området 6-9 ml per kilo idealvekt. Atelektasetendens ved slutten av ekspirasjonen hindres ved å opprettholde et positivt trykk i luftveiene under hele ekspirasjonsfasen (positivt ende-ekspiratorisk trykk, PEEP). Utbredelsen av sykdomsprosessen i lungene er viktig for valg av tidevolum og PEEP. På grunnlag av data fra en større multisenterundersøkelse (17) har flere ekspertgrupper anbefalt et standardisert tidalvolum på 6 ml per kg idealvekt for alle pasienter med akutt lungesviktsyndrom. Dokumentasjonen for at dette gir optimalt resultat for alle pasienter er imidlertid usikker (18). På grunn av store variasjoner i lungenes status hos pasienter med akutt lungesviktsyndrom bør tidevolum og PEEP tilpasses individuelt, samtidig som målsetningen bør være å velge de laveste tidevolumer som gir tilfredsstillende oksygenering og CO2 kontroll. Hos pasienter hvor atelektaser øker oksygeneringsproblemene, har ofte intermitterende hyperinflasjon av lungene (rekruttering) en gunstig effekt.
Fordi ødem er en integrert del av sykdomsbildet i første fase, ønsker man å redusere lungekartrykkene. Hvis redusert væsketilførsel nedsetter hjerte-minuttvolumet, kan arterielt O2-innhold paradoksalt nok synke når oksygenmengden i veneblodet faller selv om shuntfraksjonen er konstant eller bedres (Fig 1). Optimalisering av hjerte-minuttvolumet ved å øke hjertets endediastoliske fylling gir høyere hydrostatiske trykk i lungekarene og mer ødemdannelse. Dermed øker shuntens størrelse. Øket oksygenmengde i veneblodet kan likevel gi en høyere paO2. Ofte velger man å holde hjertets fyllingsvolum på et suboptimalt nivå, og øker kontraktiliteten i myokard med infusjoner av inotrope agenser.
Mange intervensjoner og medikamentelle behandlingsregimer har vist lovende resultater i dyreeksperimentelle studier og studier med få pasienter. Stereoidbehandling, annen antiinflammatorisk behandling, vending i mageleie, høyfrekvent ventilasjon (jetventilasjon, oscillatorisk ventilasjon), inhalasjon av nitrogenmonoksid og instillasjon av surfaktant i luftveiene er noen av de intervensjonene som har vært prøvet. Selv om flere av intervensjonene bedrer lungefunksjonen på kort sikt hos mange pasienter, har ingen av dem gitt signifikant bedring av overlevelse (hos voksne) ved utprøving i større studier (19-22) .
Enkelte pasienter utvikler katastrofal og behandlingsresistent lungesvikt og dør som følge av generalisert vevshypoksi. Ved livstruende oksygeneringssvikt økes PEEP ofte til 14-20 cm H2O, inspirasjonstiden forlenges i forhold til ekspirasjonstid (invers ratioventilasjon) og ventilasjon i mageleie prøves. Hjertets minuttvolum økes med katecholamininfusjoner for å øke oksygenmetningen i blandet venøst blod. Hvis dette ikke bedrer tilstanden prøves eventuelt høydose kortikosteroider (fortrinnsvis hos pasienter uten sepsis), eventuelt kjøles pasienten ned for å redusere organismens O2-forbruk og dermed øke oksygenmengden i blandet venøst blod. Den ultimate behandling av katastrofal lungesvikt er å oksygenere blodet utenfor kroppen (ekstrakorporal membranoksygenering, ECMO). Dette gjøres ved hjelp av en hjerte-lunge maskin hvor blod med lavt O2-innhold hentes ut fra en stor vene, oksygeneres, og pumpes tilbake til sirkulasjonen på venesiden (hvis tilfredsstillende hjertefunksjon) eller på arteriesiden (hvis hjertefunksjonen samtidig svikter). Behandlingen er svært ressurskrevende, og er derfor oftest bare aktuell hos personer hvor lungene er det eneste organet med alvorlig svikt (23).
Behandling som ikke er vist å gi økning av overlevelse er å anse som udokumentert krisebehandling. Hvis behandlerne tidligere har erfart, eller kjenner til, at enkeltpasienter har respondert positivt på slik behandling, er det likevel logisk å prøve også slike alternativer ved livstruende hypoksemi til tross for optimal respiratorinnsats. Hvis slik behandling innebærer fare for ytterligere komplikasjoner og/eller medfører svært høyt ressursforbruk bør forholdet mellom risiko og sannsynlig gevinst for pasienten overveies grundig før oppstart.
Dødeligheten av respirasjonssvikt rapporteres i området fra 33 % (akutt lungeskade) opp til 70 % (akutt lungesviktsyndrom) (24). Som for beregninger av forekomst varierer tallene betydelig fra studie til studie, og sannsynligvis av de samme årsaker. Tall som bygger på dødelighet i kontrollgruppen ved multisenterstudier (25-30%) kan være villedende (20), da pasienter med andre alvorlige risikofaktorer i tillegg til respirasjonssvikten ofte ekskluderes fra slike studier.
Hos de fleste pasienter med akutt lungesviktsyndrom er svikten reversibel hvis grunnsykdommen er det. Død som en direkte følge av lungesvikt ses imidlertid hos bare ca 10-15 % av pasientene (25). De fleste dør som en følge av at flere organer enn lungene svikter (multiorgansvikt) – summen av de patofysiologiske problemene blir for stor. Dette reflekterer antakelig at ARDS hos mange pasienter er den pulmonale manifestasjonen av en generell betennelsesprosess. Grunnsykdommen, kombinert med eventuelle preeksisterende tilleggssykdommer og alder, har derfor betydning for overlevelsen hos slike pasienter. Ved økende alder ser man også at dødeligheten hos intensivpasienter, målt ett år etter intensivoppholdet, er dobbelt så høy som dødeligheten på intensivavdelingen (26). Akutt respirasjonssvikt med alvorlig sepsis som underliggende årsak er forbundet med størst mortalitet, mens de pasientene hvor respirasjonssvikten utløses av skader (multitraumer) har best prognose (27). De som overlever kan gjenvinne en tilnærmet normal lungefunksjon.
Artikkelen er tidligere trykket i Tidsskrift for Den norske legeforening, nr 2 2010; volum 130, side 154-7, og trykkes med tillatelse.



